Mirko
BELJANSKI
Flavopereirine vs AIDS
Flavopereirine vs AIDS
The Beljanski Foundation
5 Tudor City Place #2209
New York, NY 10017
Tél: +1 (646)-808-5583
Fax: +1 (212)-308-7014
Association CIRIS
BP 9
17550-Dolus d'Oléron
Tél: +33 (0)4.74.56.58.00
Tél/Fax: +33 (0)5.46.75.39.75
http://en.wikipedia.org/wiki/Mirko_Beljanski
Mirko Beljanski

Mirko Beljanski (1923–1998) was a French-Serbian molecular biologist, and the founder of the Beljanski Foundation, a nonprofit organization focused on researching beneficial plant extracts for the treatment of cancer.[1] His later work on HIV and cancer was controversial.
Career
Beljanski was born in 1923 in Yugoslavia. He came to France to study, and lived there for the rest of his life.[2] He was married to Monique.[3] He received a PhD in 1948 from the University of Paris.[2] In 1948, he entered the CNRS and worked at the Pasteur Institute in Paris as a researcher in molecular biology.[4] He made several discoveries while studying RNA and DNA. Beljanski was made to leave the Pasteur Institute in 1978, after pursuing research against the advice of the institute, but still continued to publish scientific papers.[2] He was at the Faculty of Pharmacy of Châtenay-Malabry until his retirement in 1988.[2]
In his lifetime, Beljanski published a total of 133 scientific papers,[5] mostly written in French.[6] After his retirement he worked for another ten years in a private laboratory. During that time, he developed natural products that are said to inhibit the growth of cancer cells {The anticancer agent PB-100, selectively active on malignant cell lines, even multidrug resistant. Genetic and Molecular Biology 23, 1, 29-33 (2000), (Anti-prostate cancer activity of a ß-carboline alkaloid enriched extract from Rauwolfia vomitoria: International Journal of Oncology 29: 1065-1073, 2006), (Carboline Alkaloid–Enriched Extract from the Amazonian Rain Forest Tree Pao Pereira Suppresses Prostate Cancer Cells: Journal of the Society for Integrative Oncology, Spring 2009, Volume 7, Number 2), (Two Herbal Extracts for Protecting Prostate Cell DNA: Integrative Medicine, Vol. 9, No 2, Apr/Mat 2010). He also developed a preparation of RNA fragments that was claimed to promote the production of white blood cells and platelets (Dose escalation study of an antithrombocytopenic agent in patients with chemotherapy induced thrombocytopenia. Levin et al. BMC Cancer 2010, 10:565).
Beljanski believed he had found antivirals effective against cancer and AIDS.[4] A product made from extracts of the Brazilian paopereira tree[2] and called PB100 was claimed to be superior to AZT, which Beljanksi called "real poison".[3] Customers included François Mitterrand (via a homeopath called Philippe de Kuyper).[2][3] The French Department of Health accused him of illegally practising medicine in 1991, and he was sentenced in March 1994.[4]
Beljanski Foundation
The Beljanski foundation and CIRIS,[7] French not-for-profit organizations work together to promote knowledge about Mirko Beljanski's research. They also provided financial support for ongoing research both preclinical and clinical.[8][9]
http://www.youtube.com/watch?v=V-Tlz8jALvQ
Cancer:
Mirko Beljanski et Mitterrand
http://www.beljanski.com/engl/about/
The
Beljanski Approach
Mirko Beljanski, PhD, (1923-1998), biochemist and biologist, spent his professional career at the Pasteur Institute in Paris, studying the causes and mechanisms occurring during gene activation, cell division and tissue development in both normal and malignant states. His book, “The Regulation of DNA Replication & Transcription,” is devoted to this critical subject.
Dr. Beljanski discovered a change in the conformation of the DNA of a cell when in the presence of carcinogens (mutagens or other), pollutants, or other harmful molecules. He showed that the DNA of a cancer cell becomes unwound or destabilized, a finding that was recently confirmed in the United States. These results led him to invent the Oncotest, which allowed him to identify many molecules with carcinogenic potential. The Oncotest also permitted Dr. Beljanski to discover two specific anticancer plant extracts. John Hall, PhD, discusses this test and more about the ideas of Dr. Beljanski in the Townsend Letter article, “Destabilization of the DNA Double Helix in Cancer, Mirko Beljanski’s Theory of Carcinogenesis and Anti-Cancer Extracts”.
Monique Beljanski, widow and colleague of Dr. Mirko Belanski, summarizes different steps of this work in an article also published in the Townsend Letter, “The Beljanski Approach: Outside the Box”.
Dr. Beljanski made many more exciting discoveries and published many of his works. He was also one of the first biologists to study small RNA, which plays an important role in the growth of a cell. Certain RNA fragments stimulate the formation of white blood cells and platelets. They not only maintain normal white blood cell and platelet levels during radiation and chemotherapy, but they also preserve normal proportions among the various kinds of white blood cells, notably those that fight infections. These RNA fragments promote only the DNA of healthy cells, never that of cancer cells.
After 20 years of successful use in Europe, the Beljanski® products were brought to the US and can now be obtained from Natural Source International, Ltd.
At Columbia University, Dr. Aaron Katz’s team conducted more research on the efficacy of the plant extracts in the Beljanski® products, Rauwolfia vomitoria and Pao pereira (Pau pereira). Dr. Aaron Katz, MD and Debra Bemis, PhD have explored and confirmed the Rauwolfia vomitoria extract as an anti-cancer element and shortly thereafter have also explored and confirmed the Pao pereira as a selective anticancer extract.
A clinical trial to test the synergistic effects of Rauwolfia vomitoria and Pao pereira (Pau pereira) on men with a negative biopsy and elevated PSA was recently executed at the Columbia University, Department of Holistic Urology, under the direction of Dr. Geovanni Espinosa. The trial has now been completed and we await the final statistical results.
In the article “A Novel Combination of Plant Extracts with Promising Anti-Prostate Cancer Activity”, John Hall, PhD, Natacha Springer, Msc, and Debra Bemis, PhD explain and discuss the anticancer activity of Pao pereira and Rauwolfia vomitoria.
The prestigious publication, IMCJ (Integrative Medicine: A clinician’s journal) also published an article in their April-May 2010 issue titled, “Two Herbal Extracts for Protecting Prostate Cell DNA”
The Cancer Treatment Centers of America, who have just completed a clinical trial on the extraordinary Beljanski formula of RNA fragments, which are active during the synthesis of immune cells, published the results in the online magazine Bio Med Central under the following title: “Dose escalation study of an antithrobacytopenic agent in patients with chemotherapy induced thrombocytopenia”.
Moreover, several doctors and journalists were interested in promoting Mirko Beljanski’s work:
Michael Schachter, MD, of the Schachter Center for Complementary Medicine in Suffern, NY, has also taken an interest in the research of Dr. Beljanski and the Beljanski® products. Typically, he recommends the products as a complementary treatment for people fighting cancer. Read what Dr. Schachter says about Dr. Beljanski’s approach to combating serious afflictions.
In the Townsend Letter article “Symptoms Elimination for Prostate Disease”, Dr. Morton Walker explores the positive effects of Rauwolfia vomitoria and Pao pereira (Pau pereira), and the current research taking place in America with these extracts.
David Steinman, Publisher and Editor of The Doctor’s Prescription for Healthy Living magazine, recognized Dr. Mirko Beljanski as a green patriot. Dr. Beljanski’s research, specifically how chemicals and cancer are related, is important to the modern environmental movement. More information about the Beljanski® products, Prostabel® and Ladybel® is available thanks to Healthy Living magazine.
David Steinman has also spearheaded the publication of a book about the life and research of Mirko Beljanski titled “Extraordinary Healing: How the Discoveries of Mirko Beljanski, the World’s First Green Molecular Biologist Can Restore Your Health” by L. Stephen Coles, MD, PhD. Dr. Coles is the co-founder and the Executive Director of the Gerontology Research Group and specializes in supercentenarians and aging.
beljanski.com/engl/2012/03/new-book-cancers-cause-cancers-cure/?
Cancer's
Cause, Cancer's Cure: The Truth about Cancer, Its Causes,
Cures, and Prevention.
The scientific discoveries of Mirko Beljanski, Ph.D.
The scientific discoveries of Mirko Beljanski, Ph.D.
Antiviral
preparations
US5519028
US5519028
Flavopereirine taken alone may act as an effective active agent in the struggle against HIV viruses in mammals, including humans. More specifically, it has been found that flavopereirine is an active agent which on its own, whether in vitro or in vivo exerts a selective inhibitlye action on viral HIV infection, particularly in patients infected by HIV-1. Thus, there is provided a method for the treatment of human immunodeficiency virus comprising administering to a human patient infected with human immunodeficiency virus an effective antiviral amount of a composition consisting of flavopereirine or a pharmaceutically acceptable salt or derivative thereof and a pharmaceutically acceptable carrier. The flavopereirine, or one of its salts or other acceptable pharmaceutical derivatives, is preferably administered in solid form containing approximately 250-500 mg of flavopereirine per dose.
BACKGROUND OF THE INVENTION

Flavopereirine is an alkaloid of the beta-carboline class. It is also traditionally referred to as "H or PB 100 composition," and shows UV emission fluorescence at 250-254 and 306 nm.
Flavopereirine may be obtained from the peel of the Pao Pereira Geissospermum vellosii-Baillon Apocynaceae (see H. Rapaport et al., J. Amer. Chem. Soc. 80:1601-1608 (1958) and Beljanski et al., request for first certificate of addition #79 05853 to French patent application #78 07155 and EP-A-0 059 817.).
It is known that flavopereirine, administered intracutaneously at a dosage of 200-600 .mu.g or a dosage of 2.5-500 mg/day, preferably 30 mg/day, prevents the appearance and development of vital papules in the case of viruses of the Shope fibrome type and of vaccine.
It is also known that flavopereirine appears to act in vivo against influenza (RNA virus), and that it may moreover inhibit the multiplication of the tobacco mosaic virus (TMV) after brief contact with this virus.
European Patent Application EP-A-0 059 817 reveals that flavopereirine is active against the influenza virus; however, the half-life of a quaternary beta-carboline of this type is too short for efficient use in humans in a galenic form other than time-release capsules.
French Patent Application No. 88 15845 describes a system for improving immune defense in humans (against RNA viruses--AIDS in particular--and DNA viruses). According to this document, the inhibition of the multiplication of the viruses in question is possible only by a combination of four different substances, of which flavopereirine is only one. The pharmaceutical preparation revealed in the document must include at least one representative of each of these four categories of active substances. The flavopereirine included in this combination is administered at a dosage of 0.25 g/day, preferably orally.
SUMMARY OF THE INVENTION
It has now unexpectedly been discovered that flavopereirine taken alone may act as an effective active agent in the struggle against HIV viruses in mammals, including humans. More specifically, it has been found that flavopereirine is an active agent which on its own, whether in vitro or in vivo exerts a selective inhibitive action on viral HIV infection, particularly in patients infected by HIV-1. Thus, in accordance with the present invention there is provided a method for the treatment of human immunodeficiency virus comprising administering to a human patient infected with human immunodeficiency virus an effective antiviral amount of a composition consisting of flavopereirine or a pharmaceutically acceptable salt or derivative thereof and a pharmaceutically acceptable carrier. The flavopereirine, or one of its salts or other acceptable pharmaceutical derivatives, is preferably administered in solid form containing approximately 250-500 mg of flavopereirine per dose.
It is also an object of the present invention to provide a method of using a pharmaceutical preparation based on flavopereirine in order to provide an antiviral treatment for HIV.
A further aspect of the present invention is an article of manufacture comprising a packaging material and a pharmaceutical agent contained within said packaging material, wherein said pharmaceutical agent contains flavopereirine as the sole active ingredient, and wherein said packaging material comprises a label which indicates that the pharmaceutical agent can be used for treatment of human immunodeficiency virus.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1 is a graphic representation of the comparative counts of cells/ml as a function of the number of hours following infection, the addition of flavopereirine (labeled "H") having been made 12 hours after infection;
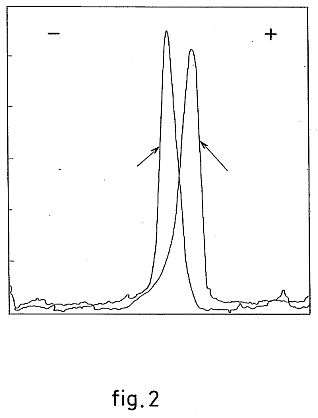
FIG. 2 represents the comparative titre of infectious units (in IU/ml) as a function of the number of hours following infection, without addition of flavopereirine and with addition at levels of 30 and 50 .mu.g/ml;
FIG. 3 represents in diagrammatic form the effect of flavopereirine on the production of interleukin-6 by human monocytes from healthy donors; and
FIG. 4 represents in diagrammatic form the effect of flavopereirine on the spontaneous production of interleukin-6 by human monocytes taken from HIV-positive patients.



Flavopereirine has the chemical formula ##STR1## For use in the present invention, flavopereirine may be prepared by the hydrolysis in 1N HCl of Geissospermum vellosii powder at 100 DEG C., followed by neutralization by KOH, extraction by ethanol and concentration by distillation. The residue of the distillation is subsequently taken up in chloroform, the excess salt is eliminated by precipitation with cold ethanol, and the residue, which contains mainly flavopereirine is concentrated. For purposes of the present invention, the flavopereirine may be used as produced by this process, or can be converted to a pharmaceutically acceptable salt or other derivative.
In accordance with the invention, flavopereirine is administered orally in the forms of capsules to patients infected with HIV at levels of 1-3 grams per day. At this level, flavopereirine is devoid of toxic or side effects in mammals, including humans. Indeed, the LD50 in Sprague-Dawley EOPS rats is 10.45 g/kg (safety limits: 9.63-11.35) when administered orally, and 2.45 g/kg (safety limits: 2.35-2.55) when administered intraperitoneally. In determining these values, when the animal died, it died within 30 to 60 minutes of oral or intraperitoneal administration by respiratory arrest. No change in mortality rate was observed during the subsequent 14 days in surviving animals.
Sub-chronic administration in male and female Sprague-Dawley rats (OPA) demonstrated the absence of toxicity in doses equal to 1/20 of the LD50 (viz. 530 mg/kg/day), or of 1/5 of the LD50 (viz. 2120 mg/kg/day). At these doses, no alterations were noted in either body weight or feeding. Neither was any alteration in the globular blood count noted, and a hepatic and renal functions remained normal. No lesions of the liver, kidneys, duodenum, myocardia, spleen, thyroid and parathyroid glands, testicles or ovaries were visible by microscope.
Flavopereirine was observed to penetrate the blood-brain barrier, as shown by the fact that in male CDI mice which received 10 mg of the flavopereirine preparation orally, the encephalic flavopereirine content was approximately 7 .mu.g (i.e., a concentration of 14 .mu.g/g, from 20 g mice with brains weighing 0.5 g.).
The effectiveness of flavopereirine in the present invention will now be demonstrated by the following non-limiting examples.
EXAMPLE 1
The destruction of HIV by flavopereirine in in vitro cell cultures without effect on normal or healthy cells was demonstrated using the H-9 colony of T4 lymphocytes obtained from Dr. R. Gallo via the Paul Ehrlich Institute (Frankfurt, Germany) and propagated by the Institut fur Medizinische Mikrobiologie und Hygiene (University of Bern, Switzerland).
HIV was obtained as HTLV-III from Dr. Gallo and propagated in the H-9 cells. The surface material produced by the infected H-9 cell culture was stored at -80 DEG C. This material had a concentration of 10@6 IU/ml at the point of use. The H-9 cells were cultivated in an RPMI 1640 medium containing 15% fetal calf serum, 0.002 .mu.m glutamine and 100 IU penicillin/ml in Falcon flasks (25 cm@3). 8 to 20 ml of the medium was used per vial. The vials were incubated at 37 DEG C. in an upright position. The culture was initiated at a concentration of 2.times.10@5 H-9 cells per ml, and divided when the number of cells reached 1.times.10@6 cells/mi. In order to test the flavopereirine in cultures of both infected and non-infected H-9 cells, microtitration plates were used. In these cases, the cultures were begun at a concentration of 6.times.10@5 H-9 cells/mi. After 23 hours incubation, 0.1 ml RPMI 1640 medium containing 10@2 IU of HIV was added to each well containing 0.1 ml of this cellular suspension. This corresponds to an infection multiplication of 1.6.times.10@4, i.e., one infectious unit per 500 cells. 16 hours after infection, 0.1 ml of RPMI 1640 medium, with or without flavopereirine, was added to the infected and non-infected wells. The flavopereirine was used at a concentration of 30 .mu.g/ml and 60 .mu.g/ml. The microtitration plates were covered with an Amersham plate cover, and incubated at 37 DEG C. Cellular counts were determined using an HIV antigen kit provided by Abbott Laboratories. For purposes of titration, a volume of 10 .mu.l (taken from each of the wells of the microtitration plate) was diluted in 1 ml of RPMI 1640 medium. Based on this stock, 8 dilutions were made in respective series of 1 to 3 and 1 to 5. A volume of 0.1 ml of each dilution was removed and added to 0.2 ml of pre-incubated culture of H-9 cells containing 5.times.10@5 cells/mi. After incubation at 37 DEG C., the presence of HIV antigens was tested using an HIV antigen kit provided by Abbott Laboratories.
The results, which are reproduced in graphic form in the attached FIG. 1, show that flavopereirine does not affect the multiplication of non-infected cells. By contrast, the quantity of infected cells is around 40% lower when the flavopereirine is present.
What was even more striking (as shown in the presentation of results in FIG. 2) was that, although there was an increase in viral particles of untreated infected cells over time, it was not possible, within the limitations of this particular test, to detect the presence of infectious units above 3000 in the series of infected cells treated with flavopereirine (30 .mu.g or 60 .mu.g). This shows that the inhibition of infection exceeds 99% at the very least.
EXAMPLE 2
An evaluation of the antiviral effect of flavopereirine was also undertaken by studying the cytopathogenic effect of the HIV virus on MT4 cells, given that a formation of syncytia was observed 4-6 days after infection by HIV-1, followed by the death of the cells.
The flavopereirine was used in the form of an alcoholic solution (40 mg in 100 .mu.l alcohol). Dilutions were made in RPMI medium at 10% of FCS, 1% of PSN and 1% of glutamine. The MT4 cells were left to pre-incubate for two hours at 37 DEG C. with a successive dilution of flavopereirine containing 3.times.10@5 cells for 10 .mu.l of flavopereirine solution. The solution was obtained by adding 100 .mu.l of a 10@-4 dilution of HIV-1 virus, producing a syncytia formation in 4-6 days. After one hour of incubation at 37 DEG C., the infected MT4 cells were washed three times with RPMI solution before being placed in culture (3.times.10@5 cells/ml in microplates with 24 wells) with the presence of different dilutions of flavopereirine. The syncytia count was taken each day in duplicate. The results are summarized in tables I, II and III below.
Table I shows cellular toxicity for 100 and 400 .mu.g/ml flavopereirine. At 50 .mu.g/ml syncytia had not formed after 7 days culture. From 10 .mu.g/ml to 100 ng/ml, syncytia was observed, as it was in the HIV-1 control,
Table II confirms the protection obtained by flavopereirine at 50 .mu.g/ml, and Table III reconfirms these results: no syncytia were formed at 60 .mu.g/ml after 7 days culture, while a few were observed after 6 days at the dosage 30 .mu.g/ml.
TABLE I
______________________________________
H d3 d4 d6 d7
400 Tox Tox
.mu.g/ml
100 Tox Tox
50 (+) (+) (+) (+) - - - -
10 + + + (+) + ++ ++ ++
1 + (+) + + ++ ++ ++/T ++/T
100 + + + + ++ ++ ++/T ++/T
ng/ml
HIV-1 + + + + ++ ++ ++ ++/T
only
MT4 - - - - - - - -
_____________________________________
TABLE II
H d3 d4 d5 d6 d7 d10
100 .mu.g/ml
Tox
Tox
50 - - - - - - - - - -
10 - - + (+)
+ + + ++ ++ ++
1 - - + + + ++ ++ ++ ++ ++/T
100 ng/ml
- (+)
+ + + + ++ ++ ++ ++
HIV-1 only
- - (+)
+ + ++ ++ ++ ++/T
++/T
__________________________________________________________________________
TABLE III
H d3 d4 d5 d6 d7
60 .mu.g/ml
- - - - - - - - - -
30 - - - - - - (+)
- ++ (+)
10 (+)
- (+)
(+)
+ (+)
++ ++ ++ ++
1 (+)
(+)
(+)
(+)
(+)
(+)
++ + ++ ++
100 ng/ml
- (+)
(+)
(+)
+ (+)
+ ++ ++ ++
HIV-1 only
(+)
(+)
(+)
(+)
+ + ++ ++ ++ ++
MT4 - - - - - - - - - -
EXAMPLE 3
To test the efficacy of flavopereirine on the infectious capability of HIV-1, two experiments were conducted. In the basic methodology of each experiment, 1 nanogram of primary isolates of HIV-1, BRE1 (from an asymptomatic patient) and TIG2 (from an AIDS patient) were inoculated with 10@6 peripheral blood mononuclear cells (PBMC) stimulated with PHA taken from five randomly-chosen HIV-negative donors. After 2 hours of incubation, the cells were rinsed twice and cultivated in 1 ml of RPMI 1640 containing 20 IU of IL-2 per ml (Boehringer Mannhelm, Germany), 2 .mu.g of Polybrene (hexadimethrine bromide) per ml (Sigma, St. Louis, Mo., USA) and 10@-7 IU of goat antiserum acting against human alpha interferon (Janssen, Beerse, Belgium) per ml. Half the culture was changed after 72 hours, and thereafter every 48 hours until the 30th day.
For the first experiment, prior to incubation of the virus in PBMC stimulated by PHA (blast cells), extracellular viral stocks were pretreated in triplicate with 30 or 60 .mu.g flavopereirine per ml for two hours. In the second experiment, blast cells were pretreated in triplicate with 30 or 60 .mu.g flavopereirine per ml for two hours, and were then rinsed twice before exposure to the viral inoculum.
The surface material of the culture was tested by an immuno-enzymatic (ELISA) assay for the production of antigen P24 (Abbott, Chicago, Ill., USA) and the optical density (OD) of the resulting color was converted into P24 concentration from the slope of a standard nomogram, as described by W. Lu and J. -M. Andrieu, Journal of Virology, 66(1): 334-340 (1992). The result of the two experiments are shown in Tables IV and V, respectively. As shown, a pretreatment of HIV-1 by flavopereirine (compound H) at a level of 30 or 60 .mu.g/ml completely prevented the infection of target PBMC by primary HIV-1 isolates taken from both symptomatic and asymptomatic patients. On the other hand, only pretreatment of target PBMC with 60 .mu.g/ml led to complete inhibition of productive viral infections.
EXAMPLE 4
To evaluate the cytotoxicity of flavopereirine in resting PBMC and in blast cells, prewashed fresh and blastic PBMC taken from five randomly-selected, healthy, HIV-negative donors were treated in triplicate with 30 or 60 .mu.g flavopereirine per ml of alcoholic solution for two hours, After washing twice, the cells were placed in culture in cellular culture medium until day 15. The viability of the cells of each group was examined by exclusion coloring with trypan blue and by quantimetric analysis. Cultures of HIV-negative PBMC without flavopereirine (compound H) were used as controls. The results are summarized in Table VI. As shown, the viability of resting PBMC was reduced significantly (p<0.05) in the group of cells treated with 60 .mu.g/ml, but this was not the case in the group treated with 30 .mu.g of compound H per ml. Viability of the blastic cells appeared to be independent of exposure to compound H.
EXAMPLE 5
To further test the inhibition of productive infection by HIV-1 through use of flavopereirine, HIV-1 was pretreated with flavopereirine (compound H) in doses of 10, 30, 60, 100, 200 .mu.g/ml and then combined with PBMC. Flavopereirine was found to inhibit infection of target PBMC by the virus in a manner dependent on the dose (see Table VII below). Doses equal to or higher than 60 .mu.g of compound H per ml appeared to represent the required concentration for complete inhibition of productive viral infections.
This experiment was repeated, but prior to the pretreatment the virus was combined with human serum. In the primary human PBMC culture system, the efficacy of flavopereirine (compound H) on the inhibition of wild HIV-1 remained unchanged when the medicine was placed for incubation in a culture medium containing 50% human serum before the inhibition experiment. (See Table VIII).
EXAMPLE 6
The cytotoxicity of flavopereirine (compound H) in human PBMC stimulated with PHA was tested in the presence and absence of human AB serum as shown in Table IX. These tests showed that the viability of blast PBMC diminished significantly (p<0.001) in the group of cells treated with 200 .mu.g/ml, but this was not the case in the groups treated with 100, 60, 30 and 10 .mu.g/ml compound H respectively. Thus, the replication of HIV-1 can be totally inhibited by a concentration of flavopereirine (60-100 .mu.g/ml) that is 2 to 4 times weaker than the cytotoxic concentrations. This effect did not appear to be influenced by the constituents of the human serum.
EXAMPLE 7
The effects of flavopereirine on the production of primary (IL-1.beta. and TNF-.alpha.) and secondary (IL-6) cytokines by monocytes was also tested. The adhesive monocytes used were taken from the blood of two types of donors:
Normal donors--voluntary blood donors who attended the blood bank of the Pitie-Salpetriere Hospital, Paris, France; and
HIV-positive donors from medical consultations. These individuals were in the early stages of HIV infection and, at the moment of taking the blood, were not undergoing any treatment. The risk factors involved were intravenous drug abuse for six of the donors, sexual transmission for three and blood transfusion (in Zaire) for one patient. No correlation was found between the risk factors and the results of the experiments.
In the case of the HIV-positive donors, a number of anomalies were found; these related mainly to the polynuclear and lymphocytic lines;
Hypersegmented or hyposegmented (Pelger type) polynuclears in four cases out of 10.
Hyperbasophilic lymphocytes were also found in four cases out of 10 (not the same cases as the above). No correlation was found between these anomalies and the results obtained in the experiments.
In immunophenotypical analysis, no expression of the differentiation antigens (CD34, CD33, CD13 and CD11b) examined was found deficient.
I. Normal donors
The ten donors in these experiments showed a considerable coherence of results;
No spontaneous production of either primary cytokines (IL-I.beta. and TNF-.alpha.) or of interleukin-6, a secondary cytokine;
The stimulation of monocytes over a period of 48 hours with interferon (1000 U/ml) did not cause any production of primary cytokines, except in donor #10 in 1L-1.beta., or of secondary cytokine, except for donors #1 and 5.
As expected, the stimulations obtained with LPS (lipopolysaccharide) and the combination interferon-.gamma.+LPS they varied in amplitude from donor to donor; they were always significant, with a synergic effect in the case of double stimulation.
Effect of product H
Product H was not used in these experiments at a rate of more than 20 .mu.g/ml water, since above this rate, net toxicity could have been produced. Four donors were previously tested with 30, 50, 100 .mu.g/ml with virtually total cytotoxicity: above 40% at 30 .mu.g/ml, and 100 .mu.g/ml above that rate (analysis of the culture surface material was not prepared).
The doses of 5, 10 and 20 .mu.g/ml were chosen from the second series of experiments onwards, after the results from the first two donors had shown that doses of less than 5 .mu.g/ml proved inactive.
Direct activity of product H
On the production of primary cytokines:
* Increase in the production of TNF-.alpha. (donor 7), in one dose only; this effect was also detected in the response to IFN-.gamma..
* Increase in the production of IL-1.beta. (donors #3, 5 and 6) with a dose effect, though this was marginal. Donor #10, however, responded very well to doses of 10 and 20 .mu.g/ml.
On the production of secondary cytokine (interleukin-6):
* No modification in response was observed.
Indirect Activity of product H
On the production of primary cytokines:
* Production of TNF-.alpha.: There was a sharp reduction in production, but solely in the case of a heavy dosage of the product (20 .beta.g/ml) (p<0.05).
* Production of IL-1.beta.: there was no significant modification of the responses to LPS or to IFN-.gamma.+LPS (p<0.05).
On the production of secondary cytokine (interleukin-6):
* There was a sharp reduction in the production of IL-6; this inhibition was never total, and, contrary to the case of TNF-.alpha., it was dose-dependent (P<0.05).
These different results are organized in diagrams 3 and 4, in which the measured doses are shown in .mu.g/ml of cytokines, and the standard deviations are not precise, since these are always 10% less than the average.
II. HIV-positive donors
The response of the monocytes taken from the various HIV-positive donors was tested in terms of spontaneous response. The quantities of cells received were low (the donors were not affected by cytapheresy), and the experiments were therefore limited by the number of cells available.
Direct activity of product H
On the production of primary cytokines.
No modification on the base production was observed, whether for TNF-.alpha. or for IL-1.beta..
On the production of secondary cytokine.
8 out of 10 donors showed a significant spontaneous response in interleukin-6.
Indirect activity of product H
Effect on the production of primary cytokines.
The effects obtained were the same as those for healthy donors, namely an almost total reduction in the production of TNF-.alpha. (only at a dosage of 20 .mu.g/ml) and an absence of any impact on the production of interleukin-1 at the three dosages employed (p<0.05).
Effect on the production of interleukin-6.
Interleukin-4 was used at the same time as flavopereirine H, since it has been shown prior to these experiments that this cytokine blocks the spontaneous production of interleukin-6 in certain HIV-positive patients.
In five cases, interleukin-9 was used for comparison, since this, too, blocks the production of IL-6 in normal monocytes stimulated by LPS.
The results obtained were as follows:
* Interleukin-4 inhibited the production of IL-6, but this inhibition was never total (p<0.05).
* Interleukin-9 partially inhibited (up to a maximum of 50%) spontaneous production; it never has a compound effect to that of interleukin-4 (indeed, in two out of five cases, it caused the neutralization of these effects; these results are not shown in the tables).
* Product H showed an inhibitory effect with a very clear dosage effect (p<0.05). However, it never led to the total inhibition of the spontaneous production of IL-6. On the contrary, in the presence of interleukin-4, an amplification effect was almost always obtained (except in case #4), with total disappearance of production at 3 .mu.g/ml of product H (and often even from 1 .mu.g/ml) (p<0.05).
In conclusion:
Product H proved toxic at dosages higher than 20 .mu.g/ml in vitro on the cells used within the framework of these experiments, viz. human monocytes taken both from healthy and from asymptomatic HIV-positive donors;
Product H proved able to modulate the production of cytokines: this was true directly for primary cytokines, though this effect was weak; indirectly, this modification was marked in the case of the production of TNF-.alpha. and IL-6, though not IL-1. Product H also inhibited the spontaneous production of IL-6 shown in some HIV-positive patients. This inhibitory effect, which in these experiments was never total, was amplified in the presence of IL-4;
The normalization of the IL-6 and TNF-.alpha. responses in the HIV positive subject, except for the inhibition of the production of IL-1, was highly significant, since product H did not modify the potential immune relations between monocytes and lymphocytes, or the majority of the inflammatory reactions necessary for survival, such as the stimulation of the stock cells of the bone marrow and the establishment of a defense reaction on the general level.
EXAMPLE 8
To test the clinical tolerance and efficacy of flavopereirine a clinical study was carried out on 24 HIV-positive patients with total T4 lymphocyte counts at absolute values ranging from 0.2-0.4.times.10@9 /1. All the patients were informed about the active ingredient being used and about the other anti-retrovirus medicines in use at the time of the study. The selection of patients was made on the basis of the absolute T4 lymphocyte count: men and women above 18 years of age, having a Karnofsky index equal to or higher than 90%, showing presence of anti-HIV I antibodies in two successive tests (ELISA method), from groups CDC 11, CDC III, CDC IV C2, CDC IV E in the CDC 87 classification; with hemoglobin higher than 100-120 g/l, neutrophile polynuclears higher than 1.5.times.10@9 /1, platelets numbering more than 80.times.10@9 /1, T4 lymphocytes numbering no more than 0.2.times.109/1 and no less than 0.4.times.10@9 /1, and the absence of anti-retroviral therapy, particularly by AZT.
20 patients were accepted on the basis of the above criteria. Before inclusion in the study, a pretherapeutic study was made for each patient: postclinical and therapeutic history, clinical examination including determination of fever, anorexia and nausea, headaches, pruritus, cough and expectoration, diarrhea, adenopathies, buccal mycoses, seborrheic dermatitis and Kaposi's lesions. A biological study was also undertaken; this included red corpuscles, platelets, lymphocytic sub-groups (CD2, CD4, VDS, CD19, CD4/8), determination of antigen P24 and microglobulin beta-2, of DHL (dehydrogenated lactate), plasmatic ferritin, ALAT (alanine-amino-transferase), ASAT (aspartate-aminotransferase) and plasmatic creatinine.
The flavopereirine (compound H) was administered in the form of 600 mg capsules,at a daily dose of 1-3 g, preferably at least around 1 g, which is generally active for one day, The average length of treatment was 43 .+-.11 weeks. Side effects were few, occurring only in the first three months. Neither blood nor renal toxicity was observed; nor was there any significant modification in ALAT or ASAT. No degradation in the CDC classification and no infections were noticed; the Karnofsky's index remained around the 100% level in all cases- Physical and professional activity on the part of the patients remained completely normal.
Immune response to the treatment was expressed mainly in a significant increase in CD4+ cells (p.<0.05), as well as in CD19+ cells (p<0.05). The negative decline in CD4+ was reversed in 18 out of 19 patients (p<0.05).
All the in vitro and in vivo results clearly indicate that the flavopereirine compound exerts a significant inhibitory effect on the viral infectional capacity of HIV, both in vitro in human cells and in vivo in HIV-1 infected patients.
In 10 of the patients treated over a year, the following significant variations were further noted:
Increase in red cells at 9 months;
Increase in hemoglobin at 9 months;
Increase in the total lymphocyte mass at 9 and 12 months;
Increase in CD2 at 9 months;
Increase in CD4 at 12 months;
Increase in CD8 at 9 months;
Increase in CD19 at 6-9 and 12 months;
Increase in microglobulin beta-2 at 3-6 and 12 months.
In practice, oral administration in solid form, such as tablets or capsules, for example, is recommended. A unitary dosage of around 250-500 mg of active ingredient is particularly appropriate.
The recommended dosage, in the light of the above results and the indications of toxicity, is around 1-3 g, which are generally active for one day (g/d) and preferably at least around 1 g/d, most profitably taken at successive intervals over the course of the day.
The dosages and/or galenic forms retained may, however, vary according to the state of the patient and the stage of viral attack being treated. Their adaptation to the specific case concerned in each particular treatment may be easily achieved by the professional on the basis of his relevant experience and, if necessary, with the assistance of routine preliminary tests. In this respect, it is particularly recommended that close attention be paid to the data provided by a pharmacokinetic study of the patient made in order to establish the half-life, of the active ingredient being administered, and, if necessary, to adapt the form of pharmaceutical preparation for administration accordingly. The latter may, for example, take the form of time-release galenic preparations.
Apart from the active ingredient or a salt or other derivative thereof, the doses for administration include at least one pharmaceutical support or vector, as well as excipients, carriers and standard perfumes and/or colorants.
TABLE IV
Pretreatment of viral inoculum with compound H (experiments in triplicate)
HIV
pretreated Post-infection production
Viral stock with H for of HIV P24 (pg/ml)
(1 ng/ml)
2 hours d3 d5 d14 d21 d30
______________________________________
HIV-1Asym.
Control 250 .+-. 25
>1500
(StockBrat)
+ 30 .mu.g/ml
+ 60 .mu.g/mi
HIV-1AIDS
Control 575 .+-. 129
>1500
(StockTigr)
+ 30 .mu.g/ml
+ 60 .mu.g/ml
Reunified peripheral blood mononuclear cells (PBMC) taken from five randomlychosen, healthy, HIVnegative donors.
TABLE V
Pretreatment of target cells with compound H (experiments in triplicate)
PBMC*
pretreated Post-infection production
Viral stock
with H for of HIV P24 (pg/ml)
(1 ng/ml)
2 hours d3 d5 d14 d21 d30
HIV-1Asmy.
Control 250 .+-. 25
>1500
(StockBrat)
+ 30 .mu.g/ml
113 .+-. 7
>1500
+ 60 .mu.g/ml
- - - - -
HIV-1AIDS
Control 575 .+-. 129
>1500
(StockTigr)
+ 30 .mu.g/ml
515 .+-. 103
>1500
+ 60 .mu.g/ml
*Reunified PBMC taken from five randomlychosen, healthy, HIVnegative donors.
TABLE VI
Cytotoxicity of compound H in human PBMC at rest and in PBMC stimulated by PHA (blastic) (experiments repeated 5 times)
Viability (%) of PBMC after Target Cells treated
exposure to compound H
cells with H (2 hrs.)
d3 d7 d11 d13 d15
PBMC* Control 97 .+-. 2
95 .+-. 3
98 .+-. 2
91 .+-. 8
85 .+-. 9
+ 30 .mu.g/ml
95 .+-. 4
93 .+-. 2
88 .+-. 7
82 .+-. 10
81 .+-. 11
+ 60 .mu.g/ml
56 .+-. 6
23 .+-. 4
17 .+-. 4
12 .+-. 5
25 .+-. 8
Blast.**
Control 86 .+-. 5
34 .+-. 6
76 .+-. 4
75 .+-. 5
68 .+-. 7
+ 30 .mu.g/ml
88 .+-. 3
35 .+-. 7
71 .+-. 3
76 .+-. 9
69 .+-. 8
+ 60 .mu.g/ml
79 .+-. 4
74 .+-. 5
70 .+-. 4
71 .+-. 8
63 .+-. 7
*Reunified PBMC taken from five randomlychosen, healthy, HIVnegative donors.
**Blastic cells stimulated with PHA taken from five randomlychosen, healthy, HIVnegative donors.
TABLE VII
Pretreatment of viral inoculum with compound H in the absence of human AB group serum (experiment repeated 5 times)
HIV pretreated
Production of P24 in HIV (pg/ml) with H for 2 hrs.
d4 d10 d14 d21
Control 510 .+-. 235
>1500
+ 200 .mu.g/ml
+ 100 .mu.g/ml
+ 60 .mu.g/ml
+ 30 .mu.g/ml
173 .+-. 102
>1500
+ 10 .mu.g/ml
388 .+-. 124
>1500
Reunified peripheral blood mononuclear cells (PBMC) taken from five randomlychosen, healthy, HIVnegative donors.
TABLE VIII
Pretreatment of viral inoculum with compound H in the presence of human AB serum (experiment repeated 3 times)
HIV With/w.out pretreated
(-/+)
with H for
50% of AB Production of P24 in HIV (pg/ml)
2 hrs. serum d4 d10 d14 d21
Control - 510 .+-. 235
>1500
+ 200 .mu.g/ml
+ - - - -
+ 100 .mu.g/ml
+ - - - -
+ 60 .mu.g/ml
+ - - - -
+ 30 .mu.g/ml
+ 275 .+-. 98
>1500
+ 10 .mu.g/ml
+ 384 .+-. 83
>1500
Reunified peripheral blood mononuclear cells (PBMC) taken from five randomlychosen, healthy, HIVnegacive donors.
TABLE IX
Cytotoxicity of compound H on human PBMC stimulated with PHA (blasts) in the presence or absence of human AB group serum (experiment repeated 5 times)
Cells With/w.out
treated (-/+) Cytotoxicity of compound H after
with H 50% of
exposure to PBMC*
(2 hrs.)
AB serum
d4 d10 d14 d21
Control - 85 .+-. 1.4**
86 .+-. 1.3
80 .+-. 2.7
77 .+-. 3.1
+ 200 .mu.g/ml
- 45 .+-. 6.3
33 .+-. 7.8
24 .+-. 7.6
15 .+-. 5.4
+ 100 .mu.g/ml
- 84 .+-. 1.6
79 .+-. 3.1
73 .+-. 5.3
74 .+-. 4.7
+ 60 .mu.g/ml
- 86 .+-. 1.5
82 .+-. 2.4
82 .+-. 2.6
80 .+-. 3.3
+ 30 .mu.g/ml
- 81 .+-. 2.8
83 .+-. 2.6
80 .+-. 3.9
78 .+-. 4.8
+ 10 .mu.g/ml
- 87 .+-. 1.2
86 .+-. 1.4
84 .+-. 1.5
81 .+-. 2.3
+ 200 .mu.g/ml
+ 37 .+-. 6.8
24 .+-. 6.6
17 .+-. 7.9
11 .+-. 5.6
+ 100 .mu.g/ml
+ 83 .+-. 2.3
80 .+-. 4.5
77 .+-. 5.6
73 .+-. 4.4
+ 60 .mu.g/ml
+ 85 .+-. 1.2
84 .+-. 1.7
82 .+-. 2.1
79 .+-. 3.5
+ 30 .mu.g/ml
+ 88 .+-. 1.3
84 .+-. 2.5
82 .+-. 3.7
81 .+-. 4.6
+ 10 .mu.g/ml
+ 87 .+-. 1.1
88 .+-. 1.4
84 .+-. 3.2
82 .+-. 3.1
*Blastic cells stimulated with PHA taken from five randomlychosen, healthy, HIVnegative donors.
**Percentage (average .+-. standard deviation) viable cells
Polyribonucleotides
capable of promoting the genesis of leucocytes and blood
platelets
US4335239
US4335239
The invention relates to new medicaments useful for the treatment of leucocyte and platelet deficiencies, which medicaments are polyribonucleotides prepared by degradation of the ribosomic ribonucleic acids extracted from micro-organisms or from animal organs and are formed of simple chains or "RNA fragments" comprising about 20 to 80 ribonucleotide units, the overall ratio of purine bases (G+A) to pyrimidine bases (C+U) being between 1.0 and 2.5. The invention also relates to a process for the preparation of these polyribonucleotides by scission of the ribosomic ribonucleic acids extracted from suitable micro-organisms, by means of a ribonuclease or of a chemicl reagent such as an alkali metal base.
This invention relates to polyribonucleotides capable of promoting generation of leucocytes and blood platelets.
Recently, in French patent application No. 74/38,768 and C. R. Acad. Sci. Paris, Series D. T. 280 (20th January 1975) pages 363-366, a process has been described for the preparation of polyribonucleotides, also called "RNA-fragments," by the action of ribonucleases which leave the guanine-adenine (G-A) sequences intact (in particular pancreatic ribonuclease) on G and A rich ribosomic ribonucleic acids extracted from bacteria or from cells of animal organs. The polyribonucleotides thus obtained comprised single-stranded chains of about 20 to 80 riboncleotide units, in which the purine bases outnumbered the pyrimidine bases, and the G-A sequence units predominated.
These polyribonucleotides have been selectively separated into various families by passing through a column filled with a molecular sieve marketed under the name "Sephadex G 25 fine" in a M/100 tris buffer of pH 7.4, elution being effected with the same buffer. The fractions separated in this way are called, in order of elution, P1, P2, P3, P4 and P5, and correspond to the peaks of the curve shown in FIG. 1 of the accompanying drawings, in which the volume of eluate is plotted as the ordinate and the absorption measured at 260 m.mu. is plotted as the abscissa.
The various families of polyribonucleotides thus obtained have been analysed spectrophotometrically, and their contents of purine bases, namely guanine (G) and adenine (A), and of pyrimidine bases, namely cytosine (C) and uracil (U) determined. The families are distinguished from one another by the amount and ratio of the bases, the families P1 and P2 being richest in purine bases G and A.
It has already been reported that the families called P1 and P2, obtained from ribosomic RNA of Escherichia coli M 500, exhibit an anti-viral activity, in particular against Shope fibroma virus, cow-pox virus and herpes virus.
The present invention provides a medicament for treating leucocyte and platelet deficiencies, comprising simple chain polyribonucleotides having 20 to 80 ribonucleotide units prepared by degradation of the ribosomic ribonucleic acids extracted from micro-organisms or animal organs, the overall ratio of purine bases to pyrimidine bases [(G+A)/(C+U)] in said polyribonucleotides being between 1.0 and 2.5.
It has now been found, according to the invention, that the polyribonucleotides, wherein the overall ratio of the purine bases to the pyrimidine bases [(G+A)/(C+U)] is between 1.0 and 2.3, promote the genesis of leucocytes and of blood platelets and are thus useful as medicaments for encouraging leucopoiesis and the formation of platelets when a deficiency occurs.
In particular, the products called P3 and P4 and obtained from ribosomic RNA of Escherichia coli M 500 Sho-R by the action of pancreatic ribonuclease both exhibit a ratio (G+A)/(C+U) of between 1.0 and 2.5, according to the invention, and are useful as "regenerator" of leucocytes and platelets.
The medicament according to the invention can be prepared in accordance with the process described in French patent application No. 74/38,768 from various sources (yeasts, bacteria and animal organs), in particular by passing E-coli M 500 Sho-R over a molecular sieve to select the fractions wherein the ratio (G+A)/(C+U) is between 1.0 and 2.5.
However, the invention also relates to an improved process for the preparation of these polyribonucleotides which makes it possible to obtain them in a simpler manner and with greatly increased yield.
The invention accordingly provides a process for the preparation of the polyribonucleotides according to the invention, wherein ribosomic ribonucleic acids extracted from a micro-organism having a ratio (G+A)/(C+U) from 1.0 to 2.5 are degraded by a ribonuclease or by a chemical reagent.
According to this improved process, the methods of culture of the bacteria, of isolating the ribosomic ribonucleic acids (r-RNA) and of preserving them may be identical to those described in French patent application No. 74/38,768, the improvement relating to:
(1) the choice of the bacterial strain or other starting material (fungi, yeasts or animal organs);
(2) the degradation agent used for the scission of the r-RNA into RNA fragments; and
(3) the resulting elimination of the need for fractionating the product on a column.
It is preferred to use a wild non-pathogenic strain of E. coli T3000 (K 12) belonging to the species which are usually hosts of the intestinal flora. In this strain, the ratio of purine bases to pyrimidine bases is about 1.0, which is less than the ratio in the strain E. coli M 500 Sho-R. However, it is possible to use r-RNA isolated from other bacterial strains, fungi, yeasts or animal organs in which the ratio of purine bases to pyrimidine bases is satisfactory.
The agents used for the degradation can be not only ribonucleases, such as pancreatic ribonuclease or a ribonuclease extracted from Neurospora crassa, but also strong bases (sodium hydroxide or potassium hydroxide), preferably at a final concentration of 0.1 N in the reaction solution.
The RNA fragments obtained in this manner from a suitable starting material exhibit an overall ratio (G+A)/(C+U) of between 1.0 and 2.5 and it is not necessary to carry out a fractionation on a column.
An example of the preparation of the fractions P3 and P4 from a strain of E. coli rendered resistant to showdomycin described by M. Beljanski et al. (C. R. Acad. Sci. Paris, Series D, 272, pages 2,107-2,110) and registered at the Centraal Bureau Voor Schimmelcultures, under No. CBS 615-74 and hereinafter referred to as E. coli M 500 Sho-R, is given below.



EXAMPLE 1
Preparation of P3 and P4 from E. coli M 500 Sho-R.
The bacteria of this strain are cultured at 37 DEG C. in a well-aerated medium either on a rich medium containing, per liter of medium, 10 g of Bacto-tryptone, 5 g of yeast extract, 5 g of sodium chloride and sodium hydroxide solution to bring the pH to 7.3, or, if it is desired at the same time to isolate the anti-viral products P1 and P2 and the products P3 and P4, on a synthetic medium containing, per liter of medium, 100 ml of monopotassium phosphate solution containing 136 g/l, 10 ml of 20% strength ammonium sulphate solution, 1 ml of 0.05% iron sulphate solution, 1 ml of magnesium sulphate solution containing 20 g/100 ml, 2 ml of vitamin B1 solution containing 0.5 part per 1,000 and potassium hydroxide solution to bring the pH to 7.2, 4 or 5 g per 1,000 of separately sterilised glucose (20% strength solution) being added to this medium after the latter has been sterilised.
At the end of the culture, the bacterial cells are collected by centrifuging and can be stored frozen. They are then homogenised in the cold in a buffer A (5 ml of 2 M tris/HCl, 30 ml of 2 M KCl, and 10 ml of a solution containing 30 g of Mg acetate/100 ml) and then ground, and their destruction is completed by ultrasonic treatment.
After dilution with a buffer B (similar to buffer A but only containing 0.1 ml of Mg acetate and 0.1 ml of mercaptoethanol), the mixture is centrifuged for 20 minutes (25 to 30,000 g) and 10 to 20 .mu.g of desoxyribonuclease per ml are then added to the supernatant liquor. The desoxyribonuclease is allowed to act for 15 minutes at 30 DEG-37 DEG C. and the mixture is then centrifuged for 20 minutes (25 to 30,000 g). The supernatant liquor is then re-centrifuged for 2 hours at 40,000 rpm in an ultracentrifuge in order to collect the ribosomes and the remove the whole of the useless RNA 4 S.
The caked ribosomes are homogenised in the presence of buffer B, 2-3 drops of 20% strength lauryl-sulphate solution are added, and the mixture is subjected to thorough mechanical stirring. The ribosomic RNA is extracted by the conventional method using phenol in the presence of buffer B, several such extractions being necessary, and a final extraction with choloroform is effected in order thoroughly to remove the phenol and proteins still present.
The aqueous phases are combined and to them is added 96 DEG strength cold alcohol containing a little KCl to assist the precipitation of the RNA. By centrifuging for 5 minutes at 5,000 g it is possible to collect the RNA which is then dialysed overnight against distilled water containing 0.1 M KCl.
In the morning, the dialysis is continued for 1 hour against distilled water only. The RNA is determined at 260 nm (U.V.) with the aid of a spectrophotometer.
The 260/280 ratio makes it possible to check whether the RNA preparation is pure. This ratio should be very close to 2.
The RNA is stored frozen or as a lyophilised powder.
The fractionation of the RNA to give polyribonucleotides (RNA fragments) is carried out as follows: 70 mg of ribosomic RNA (about 10 ml) are brought together with 0.2 ml of a solution of crystallised pancreatic ribonuclease. (The solution of pancreatic ribonuclease, containing 5 mg/ml, was beforehand boiled for 10 minutes and then cooled).
The RNA is incubated with the ribonuclease for exactly 30 minutes at 36 DEG C. (water bath). The degradation is stopped by adding an equal volume of chloroform and stirring vigorously for a few minutes. The mixture is centrifuged for 5 minutes at 5,000 g. The aqueous phase (upper phase) is removed and, for the second time, a equal volume of chloroform is added, after which the mixture is stirred and centrifuged. The aqueous phase is immediately deposited on a column of fine Sephadex G-25 equilibrated with an H2 O-0.1 M tris/HCl buffer of pH 7.4.
The RNA fragments are eluted with this same buffer.
Under these conditions, 5 peaks detectable by absorption at 260 nm appear regularly, as illustrated by the elution curve. They are called from 1 to 5 in the sequence of elution from the column.
The RNA fragments which constitute peaks 1 and 2 exhibit an anti-viral activity.
The RNA fragments constituting peaks 3 and 4 always exhibit a very spectacular activity as leucocyte and platelets regenerators and constitute the medicaments according to the invention.
The RNA fragments which constitute peak 5 were not kept.
The fractions constituting each of the peaks are combined and lyophilised. The products P1, P2, P3 and P4 are thus obtained after taking up the dry residue in the minimum amount of distilled water, treating this once, vigorously, with an equal volume of chloroform, centrifuging the mixture, dialysing the supernatant liquid for 24 hours (under oxenic conditions) against sterile distilled water and lyophilising to dryness. The products P3 and P4 are products according to the invention, as are their mixtures in any ratios.
The constitution of the RNA fragments P3 and P4, which are formed of simple chains comprising from 25 to 50 nucleotides, was studied in accordance with the technique described in French Patent Application No. 74/38,768 so as to determine their contents of purine bases and pyrimidine bases.
150 .mu.g of RNA fragments are hydrolysed for 1 hour at 100 DEG C. (in a boiling water bath). After evaporation in a dessicator, the residue is taken up in 0.02 ml of distilled water and subjected to thin layer chromatography (ecteola cellulose) in accordance with the technique described by G. R. Bjork and L. Svensson (1967, Biochim. Biophys. acta, 138, pages 430-432). The hydrolysis liberates the purine bases and the pyrimidine bases remain in the form of nucleotides.
The constituents of the RNA fragments P3 and P4 were separated by closed cell chromatography and identified in accordance with the technique described above for the RNA fragments P1 and P2.
The RNA fragment P3 consists of:
A : 29.0
G : 41.1
(G + A/C + U = 2.3)
C : 15.2
U : 15.0
The RNA fragment P4 consists of:
A : 25.6
G : 26.3
(G + A/C + U = 1.06)
C : 21.0
U : 27.1
These figures are expressed in mols per 100 mols of nucleotides analysed, using the following extension coefficients: A=13; G=12.8; C=11.5; and U=10 and correspond to the absorption maximum (see Methods in Enzymology XII. Nucleic Acides, Part A, Ed. Grossman and K. Moldave, Academic Press (1967), page 386).
The RNA fragments P3 and P4 contain no trace of DNA. This was rigorously checked by colorimetry (diphenylamine) and by enzymology (activity in the presence of DNA-polymerase).
The process for the preparation of RNA fragments according to the inventionfrom r-RNA of E. coli T 3,000, by means of various ribonucleases and by means of an alkali metal base, will now be described in Examples 2 to 4 below.
EXAMPLE 2
Degradation of r-RNA of E. coli T 3,000 by pancreatic ribonuclease A
The r-RNA were obtained from a culture of E. coil T 3,000 by a processidentical to that described in Example 1 for the r-RNA of E. coli M 500 Sho-R. The degradation of the RNA obtained is effected in accordance with the invention by means of a solution of pancreatic ribonuclease (grade A) containing 5 mg/ml, which has beforehand been heated to 100 DEG C. for 10 minutes (on a boiling water bath) and then cooled rapidly.
A mixture of r-RNA and ribonuclease is incubated at 36 DEG C., the ribonuclease concentration being the same as in Example 1, but the incubation time being shorter, namely 20 minutes (instead of 30 minutes). [If a different, more or less crystalline, sample of pancreatic ribonuclease is used, it is necessary to adapt the ribonuclease concentration or the incubation time].
The reaction is stopped by adding a solution of phenol containing 10% of distilled water (1 volume of this solution per 1 volume of reaction mixture), and this mixture is stirred vigorously so as to remove the ribonuclease. After centrifuging for 5 minutes at 10,000 rpm, the aqueous (upper) phase then has an equal volume of phenol added to it, and the mixture is stirred and then centrifuged. The operation is repeated with chloroform (using equal volumes). After separating the phases and repeating the operation two or three times, the aqueous phase is dialysed under axenic conditions for 16 hours against sterile distilled water at 4 DEG C.
The amount of non-dialysable RNA fragments is determined by absorption in the ultraviolet at 260 nm. The yield of active RNA fragments, relative to the initial amount of ribosomic RNA, varies from 50 to 60%.
The RNA fragments are stored after lyophilisation.
Electrophoresis, on acrylamide gel, of the RNA fragments reveals the presence of a single peak of RNA fragments of smaller size than that of the 4 S transferred RNA, as is shown by the attached FIG. 2, in which the absorption at 260 nm has been plotted as ordinates and the distance travelled in 1 hour 30 minutes as abscissae (a method described by M. Beljanski, P. Bourgarel and Mrs. M. Beljanski; Ann. Inst. Pasteur 1970, 118, page 253).
EXAMPLE 3
Degradation of the r-RNA of E. coli T 3,000 by ribonuclease N1
Ribonuclease N1, originating from Neurospora crassa and prepared and purified as described by K. Kasai et al., J. Bio. Chem. 1969, 66. page 389, and crystallised once, degrades the polyribonucleotide chains at the base G and its controlled action on the r-RNA makes it possible to obtain RNA fragments which are active in leucopoiesis and in the formation of blood platelets. The following conditions are used: 100 mg of r-RNA of E. coli T 3,000 dissolved in distilled water are incubated in the presence of 0.73 ml of ribonuclease N1 (initial solution of 1,000 units/2 ml). Incubation time: 30 minutes at 36 DEG C. The ribonuclease is immediately removed by the phenol and the chloroform as described in the case of ribonuclease A; the fragments are dialysed against sterile distilled water. The product obtained is lyophilised.
EXAMPLE 4
Degradation of the r-RNA of E. coli T 3,000 by sodium hydroxide solution or potassium hydroxide solution
To a solution of 7 to 10 mg of r-RNA/ml is added a solution of NaOH or KOH so as to bring the final concentration of the latter to 0.1 N.
The incubation is carried out at 36 DEG C. for 30 minutes. The mixture is immediately neutralised with an equal volume of 0.1 N HCl. The solution is dialysed against distilled water for 16 hours at 4 DEG C. The non-dialysable product obtained is lyophilised. After hydrolysis of the various RNA fragments of Examples 2, 3 and 4, the ratio of purine bases/pyrimidine bases was determined. The results are given in the table below and expressed in mols per 100 mols of nucleotides analysed.
TABLE
RNA RNA RNA
fragments fragments fragments
of of of
Bases Example 2 Example 3 Example 4
G 42.0 24.3 34.0
A 28.8 26.8 23.1
C 15.7 25.0 21.2
U 13.5 23.6 21.7
ratio (G + A)/(C + U)
2.3 1.06 1.36
No significant difference is observable between the RNA fragments obtained by the various degradation agents mentioned above and when using the conditions described above; the sizes of the fragments are virtually identical and always less than that of RNA 4 S (see FIG. 2).
Pharmacological properties
The distribution, in the organism, of RNA fragments P3 and P4 of Example 1, marked with @14 C, was studied after intravenous injection into mice or rabbits.
The RNA fragment P4 marked with @14 C essentially settles in the spleen and to a lesser extent in the liver, and is also to be found in the bone marrow. The RNA fragment P3 marked @14 C also settles and essentially in the same organs, but to a lesser degree. On killing the mice treated with these products, an increase in the volume and weight of the spleen (shown in FIGS. 3 and 4) is found solely in the case of the animals treated with P4. In FIG. 3, the weight of the spleen (in mg) has been plotted as ordinates, and in FIG. 4 the weight of the liver (in g) has been plotted as ordinates, in each case as a function of the number of days (plotted as abscissae) which elapsed after the animal was given a dose of 0.3 mg of product P3 (curves 3) of P4 (curves 4), intravenously or intraperitoneally, per 20 g of weight of the mouse. The two organs regained their normal weight after 5 to 6 weeks and radioactivity was no longer found, having undergone natural elimination.
The action on the genesis of the leucocytes and the platelets was studied in rabbits treated with methotrexate. In animals which were given methotrexate (35 mg intramuscularly in the case of rabbits), a decrease of about 30% in the leucocytes 48 hours after administration of the antimitotic agent was found. Of 3 rabbits used for the experiment, 1 rabbit was then subcutaneously given 2 mg of the mixture of P3 +P4 (in the weight ratio of 1:1); 1 rabbit was given the same dose intraperitoneally and a third rabbit was only given methotrexate (comparison). The two rabbits treated with P3 +P4 regained a virtually normal number of leucocytes in 5-7 days whilst the comparison rabbit only recovered this normal number after about 15 days.
These same rabbits were subsequently given a second dose of methotrexate (55 mg intravenously, per rabbit), on day 0 in FIG. 5.
Two days afterwards, 1 rabbit was given 5 mg of P3 +P4 (weight ratio 1/1.5) intraperitoneally, 1 rabbit was given the same dose subcutaneously and the third rabbit (the same comparison animal as in the preceding experiment) only received the methotrexate. The results of the analysis of the number of leucocytes in the blood sampled every two days for 20 days are illustrated by FIG. 5.
In FIG. 5, the number of leucocytes is plotted as ordinates as a function of the number of days (plotted as abscissae) which have elapsed after the injection of methotrexate; it is seen that the action of the methotrexate manifests itself, in the three rabbits, by a very great lowering of the number of leucocytes (comparison results: curve 1), but this lowering was less in the case of the two rabbits previously treated with P3 +P4 subcutaneously (curve 2) or intraperitoneally, than in the case of the comparison rabbit.
In the case of the rabbit which was given P3 +P4 intravenously a second time (curve 3), the leucocyte number becomes normal in 48 hours and increases for 24 or 48 hours before stabilising rapidly. In the case of the rabbit which was given P3 +P4 subcutaneously, curve 2, representing the increase in the number of leucocytes, reaches its maximum about the sixth day and then stabilises (FIG. 5). In contrast, in the case of the comparison rabbit (not treated with P3 +P4), the number of leucocytes remains at a low level and the animal does not succeed in regaining a normal number over the period of observation.
The number of red corpuscles in the rabbits treated in this way does not vary.
The pharmacological studies carried out with the RNA fragments obtained with various degradation agents in accordance with Examples 2 to 4 have shown that there is no significant difference in respect of the activity regarding leucopoiesis and regarding the formation of platelets, between the products P3 and P4 of Example 1 administered by themselves or administered as a mixture in any ratio, and each of the products of Examples 2 to 4.
A single dose of each of the products of Examples 2 to 4 (2 to 5 mg) administered intravenously to strongly and constantly immunodepressed rabbits (leucocyte number lowered by 60 to 70%) makes it possible to re-establish a normal number of leucocytes in 24-48 hours. The number of platelets can be increased by these RNA fragements by 50 to 100% relative to the number of platelets of the comparison animals. FIG. 6 illustrates the results obtained over the duration of an experiment (20 to 30 days ) with rabbits continuously treated with Endoxan (65 mg/day) and periodically treated with the RNA fragments obtained in accordance with one or other of Examples 1 to 4.
In FIG. 6, the number of days of the experiment over which the rabbits were given 65 mg of Endoxan per day has been plotted as abscissae. At the time indicated by the arrow A, the rabbits received 2 mg of RNA fragments intravenously. One of the curves of FIG. 6 was obtained by plotting the number of leucocytes as ordinates and the other curve by plotting the number of platelets as ordinates.
All routes can be used for the injection of the active RNA fragments, namely intramuscular injection (I.M.), intravenous injection (I.V.), subcutaneous injection (S.C.) and intradermal injection (I.D.); oral administration is also possible. The "response" time varies with the route chosen and depends on the dose of product. In rabbits not treated with Endoxan, which have a normal blood composition, the intravenous administration of RNA fragments does not alter the number of white corpuscles. If the dose of product is high, an increase is found, but in 24 hours the number of white corpuscles again becomes normal.
The action of various chemical and physical agents (Endoxan, Methotrexate, Thiotepa or radiation), and even a genetic deficiency causing a decrease in leucopoiesis or a decrease in platelets can, from this point of view, be counterbalanced by the action of the various abovementioned RNA fragments. The action on the genesis of platelets is less rapid than that on the white corpuscles, but does allow a large gradual recovery to take place.
The prolongation of the chemotherapy requires a repetition of the administration of the RNA fragments without causing exhaustion of the phenomenon.
Toxicology
The products P3 and P4 of Example 1 and those of Examples 2 to 4, dissolved in sterile physiological water, were administered to mice and rabbits intravenously, intraperitoneally, intramuscularly, subcutaneously and orally. Doses of 1 to 5 mg given as a single injection to mice and of 4 to 25 mg to rats, these injections being repeated on several days and for up to 15 days in succession did not make it possible to detect any toxic effect of the products.
Markedly higher doses, administered orally, also did not show a toxic effect.
Teratological studies have shown that the injection of the products according to the invention into female mice in gestation has no adverse effect either on the first generation or on subsequent generations.
The products according to the invention accordingly are perfectly harmless to animals.
Therapeutic application
Tests have shown that the products according to the invention can be administered each time one is dealing with leucopenia or a platelet deficiency, so as to bring the number of leucocytes and of platelets back to normal, without altering the remainder of the blood composition. A re-equilibration between the various types of white corpuscles takes place.
Since the products are soluble in water, they can be administered by any parenteral route, in the form of physiological solutions, or orally as any of the conventional galenical forms (potable solutions, tablets, pills and the like). The dose to be administered can vary from 10 to 20 mg depending on the nature of the illness to be treated, and the pharmaceutical compositions according to the invention contain, as the active product, at least one of the products according to the invention, at a unit dose of 2 to 100 mg, combined with a suitable pharmaceutical vehicle.
Cytodiagnostic method using alstonine as a selective marker, and diagnostic kit containing marker
US5567593
Method and reagent for detecting cancerigenic and anticancerous substances
US4264729
Anti-virus composition and its uses.
DK608089
VIRUSHEMMENDE ZUSAMMENSETZUNG UND IHRE ANWENDUNGEN.
AT94072
Biological regulator extracted from Gingko Biloba, active in different pathologies.
DK355089