Christopher ECCLES
Thermal Energy Cell
Chris Davies, Managing Director, Tel +44 (01206) 322496
The TEC is silent unlike air source heat pumps.
Significantly reduces the cost of heating with electricity.
Has the benefit of economically providing continuous heat generation.
Saves carbon emissions through increased energy output with no additional carbon dioxide generation.
Removes the need for unfriendly night time storage.
Usable with other sustainable and renewable energy sources such as photovoltaic, fuel cells, wind power, Stirling cycle engines, and tidal and hydro power etc.
Is conventionally installed and can directly replace gas based heating system.
Is an economic substitute for heating with gas or oil.
The TEC, compared with ground source heat pumps, does not require a bore hole or a large area of land.
Robert
Matthews: Daily Telegraph (UK),18
May 2003; "Take Water and Potash, Add Electricity and Get
-- A Mystery"
Christopher Eccles: WO 00/25320 --- Energy
Generation
C. Eccles: US Patent Application
20050236376 --- Energy Generation
C. Eccles: US Patent # 6,290,836 --
Electrodes
Jean-L.
Naudin: The Enhanced Cold Fusion Reactor --- http://jlnlabs.imars.com/cfr/html/cfr30.htm
Daily Telegraph (18th May 2003)
http://www.telegraph.co.uk/news/main.jhtml?xml=/ news/2003/05/18/ncell18.xml
"Take Water and Potash, Add Electricity and Get -- A Mystery"
by Robert Matthews, Science Correspondent
British researchers believe that they have made a groundbreaking scientific discovery after apparently managing to "create" energy from hydrogen atoms.
In results independently verified at Bristol University, a team from Gardner Watts - an environmental technology company based in Dedham, Essex - show a "thermal energy cell" which appears to produce hundreds of times more energy than that put into it. If the findings are correct and can be reproduced on a commercial scale, the thermal energy cell could become a feature of every home, heating water for a fraction of the cost and cutting fuel bills by at least 90 per cent.
The makers of the cell, which passes an electric current through a liquid between two electrodes, admit that they cannot explain precisely how the invention works. They insist, however, that their cell is not just a repeat of the notorious "cold fusion" debacle of the late 1980s. Then two scientists claimed to have found a way of generating nuclear energy from a similar-looking device at room temperature. The findings were widely challenged and the scientists, Martin Fleischmann and Stanley Pons, accused of incompetence, fled America to set up labs in France.
"We are absolutely not saying this is cold fusion, or that we have found a way round the law of energy conservation," said Christopher Davies, the managing director of Gardner Watts.
"What we are saying is that the device seems to tap into another, previously unrecognised source of energy."
According to Mr Davies, the cell is the product of research into the fundamental properties of hydrogen, the most common element in the universe. He argues that calculations based on quantum theory, the laws of the sub-atomic world, suggest that hydrogen can exist in a so-called metastable state that harbours a potential source of extra energy.
This theory suggests that if electricity were passed into a mixture of water and a chemical catalyst, the extra energy would be released in the form of heat.
After some experimentation, the team found that a small amount of electricity passed through a mixture of water and potassium carbonate - potash - released an astonishing amount of energy.
"It generates a lot of heat in a very small volume," said Christopher Eccles, the chief scientist at Gardner Watts.
The findings of the Gardner Watts team were tested by Dr Jason Riley of Bristol University, who found energy gains of between three and 26 times what had been put in.
In a written report, Dr Riley concluded: "Using the apparatus supplied by Gardner Watts and the procedure of analysis suggested by the company, there appears to be an energy gain in the system."
In tests performed for The Telegraph, the cell heated water to near-boiling, apparently producing more than three times the amount of energy fed into it.
Scientists admit to being astonished by the sheer size of the energy increase produced by the cell. "I've never seen a claim like this before," said Prof Stephen Smith of the physics department at Essex University.
"In the case of cold fusion, people talked about getting a 10 per cent energy gain or so, which could be explained away quite easily but this is much too big for that."
Prof Smith said he was sceptical about the theory put forward by the company. He conceded, however, that scientists had also been baffled by the source of energy driving radioactivity, as the key equation involved - Einstein's famous E = MC2 - had yet to be discovered.
According to Prof Smith, if there is a flaw in the company's claims, it lies in the measurement of the amount of electrical energy pumped into the cell. It is possible that, as sparks pass between the electrodes, there is an energy surge which would not be picked up by the instruments measuring the electrical input.
Prof Smith said: "This needs to be very carefully checked, as there could be far more energy going in than the makers think."
Prof Smith's views were echoed by Dr Riley, who said: "There's no doubt that there was a heat rise but I'd like to see a more thorough investigation of the electrical energy supplied into the cell."
While many scientists are trying to solve the mystery of the thermal energy cell, its huge commercial potential has already caused interest.
Cambridge Consultants, one of Britain's most prestigious technology consultancies, has teamed up with Mr Davies and his colleagues to develop a working prototype. "We've had a multi-disciplinary team working on this, and we're perplexed," said Duncan Bishop, head of process development at Cambridge Consultants.
"We are offering to risk-share on it, as it will need about £200,000 to prove the principle behind it."
According to the Gardner Watts team, it will take about six months to carry out tests putting the reality of the effect beyond all doubt. The company then plans to develop a prototype capable of turning less than one kilowatt of electrical power into 10 kilowatts of heat.
Mr Davies said: "The technology could be licensed by a company making household boilers for the domestic market. " He added that the plan is to have the first thermal energy cell devices on the market within two years.


How This 12-inch Miracle Tube Could Halve Heating Bills
Amazing British invention creates MORE energy than you put into it - and could soon be warming your home
It sounds too good to be true - not to mention the fact that it violates almost every known law of physics.
But British scientists claim they have invented a revolutionary device that seems to 'create' energy from virtually nothing.
Their so-called thermal energy cell could soon be fitted into ordinary homes, halving domestic heating bills and making a major contribution towards cutting carbon emissions.
Even the makers of the device are at a loss to explain exactly how it works - but sceptical independent scientists carried out their own tests and discovered that the 12in x 2in tube really does produce far more heat energy than the electrical energy put in.
The device seems to break the fundamental physical law that energy cannot be created from nothing - but researchers believe it taps into a previously unrecognised source of energy, stored at a sub-atomic level within the hydrogen atoms in water.
The system - developed by scientists at a firm called Ecowatts in a nondescript laboratory on an industrial estate at Lancing, West Sussex - involves passing an electrical current through a mixture of water, potassium carbonate (otherwise known as potash) and a secret liquid catalyst, based on chrome.
This creates a reaction that releases an incredible amount of energy compared to that put in. If the reaction takes place in a unit surrounded by water, the liquid heats up, which could form the basis for a household heating system.
If the technology can be developed on a domestic scale, it means consumers will need much less energy for heating and hot water - creating smaller bills and fewer greenhouse gases.
Jim Lyons, of the University of York, independently evaluated the system. He said: 'Let's be honest, people are generally pretty sceptical about this kind of thing. Our team was happy to take on the evaluation, even if to prove it didn't work.
'But this is a very efficient replacement for the traditional immersion heater. We have examined this interesting technology and when we got the rig operating, we were getting 150 to 200 per cent more energy out than we put in, without trying too hard.
People are sceptical - but somehow it works
'We are still not clear about the science involved here, because the physics and chemistry are very different-to everything that has gone before. Our challenge now is to study the science and how it works.'
The device has taken ten years of painstaking work by a small team at Ecowatts' tiny red-brick laboratory, and bosses predict a household version of their device will be ready to go on sale within the next 18 months.
The project, which has cost the company £1.4million, has the backing of the Department of Trade and Industry, which is keen to help poorer families without traditional central heating or who cannot afford rocketing fuel bills.
Ecowatts says the device will cost between £1,500 and £2,000, in line with the price of traditional systems.
The development of the groundbreaking technology results from a chance meeting between Ecowatts chairman Chris Davies, his wife Jane and an Irish inventor, Christopher Eccles, while the couple were on holiday near Shannon in 1998.
After the inventor showed the couple his laboratory experiments, Mrs Davies, immediately signed a £20,000 cheque on the bonnet of her car and handed it over to Mr Eccles.
He later became chief scientist of Ecowatts' parent company Gardner Watts, but has since left after 'falling out' with the company, according to insiders. Sadly, Mrs Davies died three years ago, so she will be unable to share in the success of her husband's development of the idea.
Mr Davies, now 75, of Dedham, Essex, was unavailable for comment last night.
But Ecowatts chief executive Paul Calver said: 'When Jane Davies whipped out her cheque book, it turned out to be a very good investment indeed.
'She and Chris were always interested in ecology and now it looks as if our heat exchanger system is ready to go on sale soon. We're producing a device in the next nine months to heat radiators.
'Most British homes rely on gas, and the Government has admitted there is a problem getting a substitute. Our device will help solve that.'
Sustainable energy expert Professor Saffa Riffat, of Nottingham University, has also led a team investigating the system.
He said: 'The concept is very interesting and it could be a major breakthrough, but more tests are required. We will be doing further checks.'
Engadget.com
Nov 10th 2007
EcoWatts "Free Energy" Device Rebuffed, BBC Falls For It.
Posted by Conrad Quilty-Harper
EcoWatts and its fake free energy gadget is back in the limelight again, with the BBC Breakfast Show falling hook, line, and sinker in an interview with the company's "CEO" Paul Calver. Calver stated that "we're still getting to the question of why it works," explaining to a BBC presenter his bewilderment at his very own creation. The response from the interviewer? "The point is it does." Unfortunately, the point is that it almost certainly doesn't. Ben Goldacre used his excellent Bad Science Guardian column this week to dig up some dirt on the dodgy company, and managed to find a scientist who gave his stamp of approval to a similar free energy gadget four years back: "Using the apparatus provided, it's true, this scientist could get incredible results: the meters would read zero, and yet water would boil in around five minutes. Because the meters provided weren't working." The company that provided this former gadget along with the "broken" meters? EcoWatts.
WO 00/25320
"Energy Generation"
Abstract
Methods and apparatus are described for releasing energy from hydrogen and/or deuterium atoms. An electrolyte is provided which has a catalyst therein suitable for initiating transitions of hydrogen and/or deuterium atoms in the electrolyte to a sub-ground energy state. A plasma discharge is generated in the electrolyte to release energy by fusing the atoms together.
Description
The present invention relates to the generation of electricity, and more particularly to the release of hydrogen and fusion of light atomic nuclei.
Normally, fusion processes are able to be initiated only at extremely high temperatures, as found in the vicinity of a nuclear fusion (uranium or plutonium) detonation. This is the principle of most thermonuclear bombs. Such a release of energy is impractical as a means of providing the power to generate electricity and heat for distribution, as it occurs too rapidly with too high a magnitude for it to be manageable.
In recent years, many attempts have been made to initiate controlled fusion processes at high temperatures by the enclosure of a region of plasma-discharge within a confined space, such as a toroidal chamber, using electromagnetic restraint. Such attempts have met with little commercial success to date as systems which employ such a technique have so far consumed more energy than they have produced and are not continuous processes.
Another approach which has been attempted in order to achieve fusion of light nuclei has been the so-called "cold fusion" technique, in which deuterium atoms have been induced to tunnel into the crystal lattice of a metal such as palladium during electrolysis. It is claimed that the atoms are forced together in the lattice, overcoming the repulsive electrostatic force. However, no clear and unambiguous demonstration of successful cold fusion has yet been presented publicly.
The present invention provides a method of releasing energy comprising the steps of providing an electrolyte having a catalyst therein, the catalyst being suitable for initiating transitions of hydrogen and/or deuterium atoms in the electrolyte to a sub-ground energy state, and generating a plasma discharge in the electrolyte. The applicants have determined that this method generates substantially more energy than the power input used to generate the plasma, whilst doing so in a controllable manner.
Preferably, the plasma discharge is generated by applying a voltage across electrodes in the electrolyte and an intermittent voltage has proved particularly useful in increasing the level of energy generation. It also provides a means of controlling the process to maintain a consistent level of energy production over a significant period of time.
The application of a voltage higher than necessary to generate plasma also is beneficial to the process and will be typically in the range of 50 V to 20,000 V and preferably between 300 V and 2,000 V, but may be higher than 20,000 V, whereas in conventional electrolysis techniques low voltages of about 3 volts are used and applied continuously across the electrodes.
The applied voltage may be DC or provided at a switching frequency of up to 100 KHz. The duty cycle of the applied voltage is preferably in the range of 0.5 to 0.001, but may be even lower than 0.001. During the pulse period a monomolecular layer of metal hydride may be formed at the cathode-Helmholtz layer interface and subsequently decays to form gas in the nascent state comprising comprising monoatomic hydrogen and/or deuterium. The waveform of the applied voltage may be substantially square shaped. Whilst application of DC to the electrode does produce the metal hydride and monoatomic hydrogen and/or deuterium, the use of a pulsed voltage has been found to be more efficient as most dissociation of the hydride then occurs between the pulses.
In applications where the electrolyte is flowed past the electrodes it may be preferable to use two separate cathodes, the first of which will be engineered to optimize production of H/D atoms and the second of which will provide the plasma discharge. In this instance the direction of flow of the electrolyte is from first to second cathode. The design of the apparatus seeks to direct the flow of electrolyte to maximize contact of monoatomic H or D atoms with the plasma. The characteristics and magnitude of the voltages applied to each cathode are preferably similar, but may have different duty periods.
In a preferred embodiment, the cathode design and applied voltage are such as to provide a current density of 400,000 amps per square meter or even greater. More preferably, the current density at the cathode is 50,000 amps per square meter or above.
In carrying out a preferred method in accordance with the invention, it has been found that the process may be assisted by initial heating of the electrolyte, which may be water or a salt solution, prior to applying electrical input to the vessel. A temperature in the range of 40 to 100 C, or more preferably 40° to 80° C, has been found to be particularly beneficial.
The ratio of water to deuterium oxide (D2)) in the electrolyte may be varied to control the energy generation. In some circumstances it may be preferable to use "light" water H2O alone and in others to use D2O alone. Additionally, the amount of catalyst added to the electrolyte may be varied as a controlling factor and preferably lies in the range of 1 to 20 mMol.
In preferred embodiments, the method includes the step of generating a magnetic field in the region of the electrodes. The intensity and/or frequency of the current used to generate the field may be adjusted to move the plasma discharge away from the electrode from which it is struck in order to minimize erosion and extend the operating life of the system. Only slight separation may be required to achieve this effect.
In further preferred embodiments, the heat generated by the process may be removed and utilized by way of a number of known and proven technologies including the circulation of the electrolyte through a heat exchanger, or using heat pipes to produce heating, or alternatively to produce electricity using a pressurized steam cycle or a low-boiling-point fluid turbine cycle, or by other means.
The present invention further provides apparatus for carrying out methods discloded herein comprising an anode, first and second cathodes, a reaction vessel having an inlet and an outlet, means for feeding an electrolyte through the vessel from its inlet to its outlet, the electrolyte having a catalyst therein suitable for initiating transitions of H and/or D atoms in the electrolyte to a sub-ground energy state, menas for applying a voltage across the anode and the first cathode to form H and/or D atoms, and means for applying a voltage across the anode and second cathode to generate a plasma discharge from the first cathode.
During the methods described herein, atoms of H and/or D are believed to undergo a fundamental change in their structure by exchange of photons with salts in solution. The applicants believe that this change, and the observed phenomena, can be explained as set out below.
It is well known that a system comprising a spherical shell of charge (the electron path) located around an atomic nucleus constitutes a resonant cavity. Resonant systems act as the repository of photon energy of discrete frequencies. The absorption of photon energy by a resonant system excites the system to a higher-energy state. For any spherical resonant cavity, the relationship between a permitted radius and the wavelength of the absorbed photon is:
2 pi r = n lambda
(pi = 3.14...)
where n is an integer
and lambda is the wavelength
For non-radiating or stable states, the relationship between the electron wavelength and the allowed radii is:
(2) 2 pi [nr1] = 2 pi r(n) = n lambda(1) = lambda(n)
where n = 1
or n = 2, 3, 4...
or n = 1/2, 1/3, 1/4...
and lambda(1)
= the allowed wavelength for n = 1
r(1) = the allowed radius for n = 1
In a hydrogen atom (and the following applies equally to a D atom), the ground state electron-path radius can be defined as r(0). There is normally no spontaneous photon emission from a ground state atom and thus there must be a balance between the centripetal and the electric forces present. Thus:
(3) [ m(e) . v12 ] / r(0)= Ze2 / ( 4 pi . epsilon(0) . r(0)2 )
where m(e)
= electron rest mass
v1 = ground state electron velocity
e = elementary charge
epsilon(0) = electric constant (sometimes referred
to as the permitivity of free space)
Z = atomic number (for H, 1)
Looking first at the excited (higher energy) states, where the hydrogen atom has absorbed photons of discrete wavelength/frequency (and hence energy), the system is again stable and normally non-radiating, and to maintain force balance, the effective nuclear charge becomes Zeff = Z/n, and the balance equation becomes:
(4) [ m(e) . vn2 ] / nr(0) = [ e2 / n ]
where n = integer
value of excited state (1, 2, 3...)
vn = electron velocity in the nth excited state.
He absorption of radiation by an atom thus results in an excited state which may decay to ground state, spontaneously, or be triggered to do so, resulting in the re-release of a quantum of energy in the form of a photon. In any system consisting of a large number of atoms, transitions between states are occurring continuously and randomly and this activity gives rise to the observable spectra of emitted radiation from H.
Each value of n corresponds to a transition which is permitted to occur when a resonant photon is absorbed by the atom. Integer values of n represent the absorption of energy by the atom.
Fractional values of are allowed by the relationship between the standing wavelength of the electron and the radius of the electron-path, given by (2), above. To maintain force balance, transitions involving fractional values for n must effectively increase the nuclear charge Z to a figure Zeff, and reduce the radius of the electron-path accordingly. This is equivalent to the atom emitting a photon of energy while in the accepted ground state, effecting a transition to a sub-ground state. Because the accepted ground state is a very stable one, such transitions are rarely encountered but the applicants have discovered that they can be induced if the atom is in close proximity to another system which acts as a "receptor-site" for the exact energy quantum required to effect the transition.
The emission of energy by a hydrogen atom in this way is not limited to a single transition "down" from ground state, but can occur repetitively and, possibly, transitions from 1/3, 1/4, 1/5, etc. states may occur as a single event if the energy balance of the atom and the catalytic system is favorable. Of course, the usual uncertainty principles forbid the determination of the behavior of any individual atom, but statistical rules govern the properties of any macroscopic (>109) quanta system.
When a "ground-state" hydrogen atom emits a photon of around 27 eV, the transition occurs to the ao/2state as demonstrated above and the effective nuclear charge increases to +2e. A new electron path radius is reduced. The potential energy of the atom in its reduced-radius state is given by
V = -{ Z(eff)e2 / [ 4 pi epsilon(o) (a(o) / 2)]} = - { 4 x 27.178} = -108.7 eV
The kinetic energy, T, of the reduced electron path is given by
T = - [ V / 2 ] = 54.35 eV
Similarly, it can be seen that the kinetic energy of the ground sate electron path is about 13.6 eV. Thus there is a net change in energy of about 41 eV for the transition:
H{ Z(eff) = 1 ; r = a(o) } to H{ Z(eff) = 2 ; r = a(o) / 2 ]
That is to say, of this 41 eV, about 27 eV is emitted as the catalytic transfer of energy occurs, and the remaining 14 eV is emitted on restabilization to the force balance.
The radial "ground-state" can be considered as a superposition of Fourier components. If integral Fourier components of energy equal to m x 27.2 eV are removed, the positive electric path inside the electron path radius increases by
(m) x 1.602 x 10-19C
The resultant electric field is a time-harmonic solution of the Laplace equations in spherical coordinates. In the case of the reduced-radius H atom, the radius at which force balance and the non-radiative condition are achieved is given by
R(m) = a(o) / [m+1 ]
Where m is an integer.
From the energy change equations given above, it will be appreciated that, in decaying to this radius from the so-called "ground-state", the atom emits a total energy equal to
(5) [ ( m + 1 )2 - 12 ] x 13.59 eV
The applicants have found that such energy emissions as take place according to (5), above, only appear to occur when the H or D is found in the monoatomic (or so-called "nascent") state. Molecular H might be made to behave similarly, but the transition is more difficult to achieve owing to the higher energies involved.
In order to achieve the transition in monoatomic H or D, it is necessary to accumulate the molecular form in the gas phase on a substrate such as nickel (Ni) or tungsten (W) which favors the dissociation of the molecule. As well as being dissociated into the monoatomic form, the H or D should be bound to the catalytic system to initiate the reaction. The preferred method of achieving this is by electrolysis using cathode material which favors dissociation.
The applicants have discovered that the catalytic systems which encourage transitions to sub-ground-state energies are those which offer a near-perfect energy couple to the [ m x 27.2 ] eV needed to "flip" the atom of H or D. It appears from experiment that the effective sink of energy provided by the catalyst need not be precisely equal to that emitted by the atom. Successful transitions have been achieved when there is an error of a s much as +- 2% between the energy emitted by the atom and that absorbed by the catalytic system. One possible explanation for this is that, in a macroscopic sized system, although the transitions are initiated by a close match in energy level, such discrepancies as arise are manifested as an overall loss or gain in the kinetic energies of the recipient ionic systems. It is thought that spectroscopic analysis of active H or D catalytic systems may provide evidence of this.
One catalyst that has been found to initiate the transition to the ao/n state is rubidium in the Rb+ ionic species. If a salt of Rb, such as the carbonate Rb2CO3 is dissolved in either water or deuterium oxide (heavy water), a substantial dissociation into Rb+ and (CO3)2- ions takes place. If the Rb+ ions are bound closely to monoatomic H or D, the transition to the ao/n state is encouraged by the removal of a further electron from the Rb ion, by provision of its second ioization energy of about 27.28eV. Thus:
Rb+ +H { a(o) / p ] +27.28 eV -->
Rb2+ + e- +H { a(o) / [ p = 1] } + { [ ( p + 1 )2 -p2 ] x 13.59 } eV
Where p represents an integral number of such transitions for any given H and D atom and by spontaneous re-association:
Rb2+ + e- = Rb+ +27.28 eV
Thus, the Rb catalyst remains unchanged in the reaction and there is a net yield of energy per transition.
Other catalytic systems can be used which have ionization energies approximating to [ m x 27.2 ] eV, such as titanium in the form of Ti2+ ions and potassium in the form of K+ ions.
The applicants believe that the above explanation is consistent with currently accepted quantum theory as discussed below.
Commencing with the equations of Rydberg and Scroedinger it can be shown that fractional numbers for the quantum theory energy states in H yield possible transitions which result in emissions at frequencies which are in accord with observed UV and X-ray spectra. It is therefore possible that the conditions conducive to initiating such transitions may be artificially reproduced in the laboratory under certain circumstances.
The Rydberg formula for the frequency of emitted radiation from a transition in monoatomic H is:
V = R(h)c( 1 / n(2)2 - 1 / n(1)2 )
Where:
V is the frequency
of the emitted photon
R(h) is the Rydberg constant, 1.097373 c 107
m-1
C is the speed of light in vacuo, 2.997 x 103 ms-1
and
n(1), n(2) are the transition states.
It can be seen from the above that, if the resultant energy state of the H atom is that which requires n(2) to be equal to 1/2 , emissions will occur which are of higher frequency than the observed Lyman 2-1 transition in the ultra-violet at 2.467 x 1o15 Hz (about 121 nm). There is, indeed, an observed emission at a wavelength of about 30.8 nm, which appears to be confirmed by recent studies of galactic cluster emissions by Bohringer, et al. (Scientific American, January 1999) and it is difficult for the inventor to conceive of any other quantum-mechanical event which would give rise to such an emission, other than a transition, in accord with the above theory, from 1 to 1/2 in nascent H.
As can be seen from the above use of the standard Rydbberg equation, such behavior of H in the monoatomic state views the conventional H "ground-state" as one of many stable electronically-preferred states for single H atoms.
To summarize, a proliferation of H or D atoms is produced which may have had significantly diminished electron-path-radii by virtue of exchange of photons with their envronment. These atoms appear to be relatively unreactive chemically and appear not to readily take the molecular form H-H or D-D. This is a fortunate property which has significance and enables fusion pathways, as described below.
The fusion of light nuclei, H and D, to form heavier elements such as He is one which has traditionally been encouraged by subjecting the reactants to extremes of temperature and pressure. This has been necessary because there is a large electric charge barrier to overcome in order to bring nuclei close enough for fusion to occur.
Using atoms with a diminished electron path radius, adjacent nuclei may experience a corresponding reduction in electric barrier and internuclear separations may become smaller. With reductions in internuclear separation, fusion processes become more probable, and more easily occasioned.
There are two principle fusion pathways for D atoms. The first is:
1D2 + 1D2 = 2He3 + 1n0
where two D nuclei fuse to produce an isotope of He and a free neutron, which subsequently decays (half-life 6.48 x 102 S), with emission of a beta particle of medium energy (about 0.8 MeV), and a type of neutrino, to become a stable proton.
The second is:
1D2 + 1D2 = 1T3 + 1H1
where the two D nuclei fuse to produce the isotope of H known as tritium (T) and a free stable proton. The tritium eventually decays (half-life 12.3 years), with emission of a beta particle of very low energy (about 0.018 MeV), to become 2He3.
Of the two, the second fusion path is preferred for the peaceful exploitation of its energy yield, because the fusion products are relatively harmless on production, and decay to completely innocuous species within a short time, emitting radiation which can be effectively shielded by a thin sheet of aluminum foil or by 10 mm of acrylic plastic, for example.
When D nuclei are forced together under high temperature and pressure conditions (as in a thermonuclear bomb), there is a greater than 50% probability for the first pathway to be the dominant one. This is because the high temperature process takes no account of nuclear alignment at the point of fusion. It is actually a matter of focusing nuclei together indiscriminately and hoping that enough fuse to produce an explosion. The applicants believe, however, in accord with established theory, that it is the alignment of the nuclei with respect to the charges in each nucleus which ultimately determines the favorable fusion path.
In order to achieve a higher probability for the second, less hazardous pathway, the approaching nuclei need to have time to align electrostatically such that the proton-proton separation is at a maximum. This can only be achieved at far lower energies than those found in a thermonuclear bomb. By the use of entities with diminished electron-path-radii, and correspondingly potentially smaller internuclear distances, fusion can be initiated at lower temperatures (and consequently lower energies), allowing for the charge-related alignment necessary to achieve a high probability for the second, tritium-forming, pathway. By introducing D of diminished electron-path-radius into the plasma discharge which is confined within the water in the vessel itself, fusion may be initiated. Temperatures of the order of 6000° K are obtained within certain plasma discharges and this, coupled with multiple quantum transitions to produce D of diminished electron-path-radius, produces a substantial yield of energy from the two-stage process.
Another possible but less likely fusion pathway for hydrogen atoms is:
1H1 + 1H1 = 1D2 + Beta+ + tau
whereby Beta+ is produced as one of the products.
Embodiments of the invention will now be described by way of example and with reference to the accompanying schematic drawings, wherein:
Figure 1 shows an apparatus for carrying out a method in accordance with the invention on a relatively small scale;

Figure 2 shows a system for operating and measuring the performance of the apparatus of Figure 1;

Figure 3 shows a circuit diagram high voltage, high frequency switching circuit for the system of Figure 2;

Figure 4 shows an apparatus for carrying out a method in accordance with the invention on a larger scale than that of the Figure 1 apparatus; and

Figure 5 shows a further apparatus for carrying out a method of the invention which includes two cathodes.

The apparatus of Figure 1 enables the generation of energy according to the principles of the invention in the laboratory. Any risk of thermal runaway is minimized whilst demonstrating that the level of energy release from the two stages is far in excess of that which would result from any purely chemical or electrochemical activity. It also enables easy calorimetry, safe ducting away of off-gases, and of subsequent extraction of liquid for titration (to demonstrate that no chemical action takes place during the operation of the apparatus).
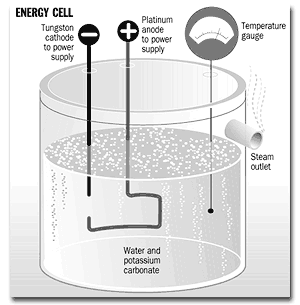
A 250 ml beaker is provided with a glass quilt or expanded polystyrene surround 6 to act as insulation. This can include an inspection cut-out so that the area around the cathode 9 can be observed from outside. The beaker contains 200 ml of water, into which is dissolved a small quantity of potassium carbonate so as to give a solution of approximately 2 mMol strength. A platinum wire 1 is earthed to the laboratory reference ground plane. The anode 10, a sheet of platinum foil of approximately 10 mm2 in area, is attached to this wire by mechanical crimping. A digital thermometer 2 is inserted into the liquid in the vessel. A 0.25 mm diameter tungsten wire cathode 9 is sheathed in borosilicate glass or ceramic tube 4 and sealed at the end immersed in the electrolyte so as to expose 10 mm to 20 mm of wire in contact with the liquid. The entire assembly of lead wires and the thermometer is carried by an acrylic plate 5 which enables of easy dismantling and inspection of the apparatus.
A supply of up to 360 volts DC, capable of supplying up to 2 amperes, is arranged external to the described apparatus. The positive terminal of this supply is connected to one pole of an isolated high-voltage switching unit. The other pole of the switch is connected to the tungsten wire cathode 9 externally of the apparatus.
To operate the apparatus, the solution 8 is initially brought up to between 40° C and 80° C either by preheating outside the apparatus or by passing power through a heating element in the solution (not shown). When the solution is between these temperatures it is either transferred to the above apparatus or, if a heating element is used, this is turned off.
With all connections made as described, the switch is set to operate at a duty cycle of 1% and a pulse repetition frequency of 100 Hz. It will be seen through the inspection cut-out that an intense plasma-arc is intermittently struck under the water at or near the cathode. If equipment is available to monitor the current drawn, it will be seen that the system consumes in the region of 1 watt when the switching circuit is operating. It will be seen by the rapid rise in temperature in the apparatus that far more energy is being released than can be accounted for by the electrical input. As a comparison, a heater element can be substituted for the electrodes and operated at 1 watt and the effects observed. There is really no need for sophisticated calorimetry to verify that large quantities of energy are being released close to the cathode of the equipment, such is the magnitude of the reaction for the process, as compared to a test with a resistive heating element of the same input power.
The data obtained from a representative one-hour session with this apparatus is shown in Table 1, below:
Table 1
Pre-Run Measurements :
Commencing volume of
electrolyte = 0.200 liter
Commencing temperature of cell = 39.200° C
Laboratory ambient temperature = 20.500° C
Specific heat capacity of vessel = 70.300 J. °C-1
Specific heat capacity of electrolyte = 4180.000 J. I-1
°C-1
Steady RMS voltage = 4.0 volts
Steady RMS current = 0.067 amps
Post-Run Results :
Duration of input =
3600 seconds
Final volume of electrolyte = 0.180 liter
Final temperature of cell = 93.6° C
Steady RMS voltage = 6.7 volts
Steady RMS current = 0.122 amps
Time-averaged power in = 0.506 watts
Results Summary :
Vessel gain =
3824.320 Joules
Electrolyte gain = 43181.740 Joules
Radiated power = 38681.030 Joules
Evaporated loss = 48509.240 Joules
Total Energy In =
1820.070 Joules
Total Energy Output = 134196.300 Joules
It can be seen from this table that the total energy input during this test was measured at 1820 J and, taking as a rough guideline that 200 ml of water requires the input of 838 J of energy to raise it by 1° C, then by direct heating the water would be expected to rise by some 2° C, bearing in mind radiative losses. In fact, during the experiment the water temperature was raised from 39.2° C to 93.6° C and considerable steam was also liberated. Furthermore, the calculated energy output of 134196 J does not take account of secondary effects such as light-energy output and Faradaic electrolysis
A system suitable for operating the apparatus of Figure 1 is illustrated in block diagram in Figure 2. A pulse generator 20 supplies a variable duty-cycle pulse waveform to to a high-voltage switch unit 22. The pulse waveform may be monitored on an oscilloscope 24 and its repetition frequency is displayed on a first frequency counter 26. A second frequency counter 28 is provided to monitor the clock speed of the switch unit 22. Power supply 30 is operable to apply a voltage between 0 and 360 V to an electrode of the apparatus 12, shown in Figure 1. The voltage level may be read from a digital multimeter 32. The RMS voltage across the electrodes 9 and 10 is indicated on a multimeter 34 and the RMS current passing between the electrodes is shown on another multimeter 36, by measuring the voltages developed across a 1 ohm resistor 37. The temperature in the apparatus 12 is indicated on a dip temperature probe 38. The switch unit 22 may be bypassed by a push button switch 39 to apply a constant voltage across the electrodes.
A circuit diagram of the switch unit 22 is shown in Figure 3. In the system of Figure 2, input 40 is connected to the output of pulse generator 20. The output 42 of the switch unit is connected to the cathode of the apparatus 12. Two NAND gates 44 and 46 are two fourths of a Schmitt-trigger 2 input NAND gate chip type 4093. NAND gate 44 operates as an astable multivibrator, with its repetition frequency set by a preset resistor 45. The output of gate 44 is fed to one in-out of NAND gate 46, the other input forming circuit input 40. The output of NAND gate 46 is connected to a three-transistor amplifier consisting of transistors 48, 50 and 52. The amplifier is in turn connected to one end of the primary of a transformer 54, the other end being connected to earth. The transformer output is fed to a bridge rectifier formed from diodes 56, 58, 60 and 62.
The rectifier output is fed via a resistor 64 to the gate of an insulated gate bipolar transistor 6 (IGBT). The load of the apparatus 12 is connected in the drain circuit of the IGBT. A 15 kV diode 68 is connected between the drain and the source of the IGBT 66 to protect the IGBT from the sizeable EMI emissions from the plasma discharges in the apparatus 12 and avoids damage to this sensitive semiconductor. A further diode 70 is provided between the drain of the IGBT and the circuit output 42 to act as an EMI blocker in a similar way. A standard 20 mm 5 amp quick-blow fuse 69 is connected between the source of the IGBT and ground in order to protect the device against over-current.
The operation of the circuit of Figure 3 is as follows. The repetition frequency in NAND gate 44 is preferably set to between 4 and 6 MHz. Pulse generator 20 is adjusted to set the duty of the switching. On receipt of an external pulse from the generator, NAND gate 46 passes a packet of 4 to 6 MHz square waves to the amplifier. The amplifier has considerable current gain and enable the primary of the transformer 54 to be driven resonantly with the RC circuit formed by capacitor 72 and resistor 74 which are connected in parallel therewith. The transformer 54 has a step-up ratio of 2:1 and a 4 to 6 MHz signal of approximately 19 volts appears across the bridge rectifier. The impedance of the rectifier output is essentially determined by a parallel resistor 76, such that the switch-on and switch-off time of the IGBT 66 is very fast. Thus, there is never a point in the operation of the device when it is dissipating any measurable power. The load of the apparatus 12 is placed in the drain circuit of the IGBT, which is therefore operating in "common-source" mode to ensure that its source terminal never rises above the high-side ground potential. This, again, is a configuration which uses excess input power. This circuit ensures a rise time of the switched waveform which isles than 10 nS and a fall time which can be as low as 30 nS at modest supply voltages.
Preferred component values and types for the circuit of Figure 3 are as follows:
Transistors 4, 50 =
2N 3649
Transistor 52 = 2N 3645
Diodes 56, 58, 60, 62 = BAT85 Schottky
Transformer 54 = RS195-460
IGBT 66 = GT8Q101
Diode 68 = 15 kV EHT
Diode 70 = 1N1198A
Resistor 47 = 1.8 kOhm
Resistor 51 = 33 Ohm
Resistor 53 = 220 Ohm
Resistor 74 = 56 Ohm
Resistor 76 = 560 Ohm
Resistor 64 = 56 Ohm
Capacitor 49 = 10 pF
Capacitor 55 = 33 nF
Capacitor 72 = 22 pF
A second apparatus for carrying out the invention is illustrated in Figure 4. This apparatus comprises a tubular chamber 80, which may be constructed from a nonmagnetic metal or metal alloy material such as, but not exclusively, aluminum or Duralumin, or alternatively may be constructed from a non-permeable ceramic material or from borosilicate glass. The tubular chamber 80 is constructed in flanged form to allow of its incorporation into a system of pipework via flanges 82 and 84 and gaskets 86. Entering the chamber 80 are two electrodes, the cathode 88 being shaped so as to present a circular plate opposite the cathode 88. The distance between the cathode tip and the anode plate should be approximately equal to the radius of the chamber 80. The cathode may be constructed from tungsten, zirconium, stainless steel, nickel or tantalum, or any other metallic or conductive ceramic material which may contribute to, or occasion, the dissociative process described above. The anode may be constructed from platinum, palladium, rhodium or any other inert material which does not undergo any significant level of chemical interaction with the electrolyte.
Surrounding the chamber 80, and concentric with it, is a winding 98 of enameled copper or silver wire of diameter 0.1 to 0.8 mm consisting of up to several thousand turns of the wire. The purpose of this winding is to create an axial magnetic field inside the chamber 80.
Electrolyte comprising deuterium oxide, in combination with ordinary "light" water in varying proportions, and containing high-molarity salts of, but not exclusively of, potassium, rubidium or lithium, or combinations of such salts, is pumped through the chamber 80, in a direction such that the anode is downstream of the cathode.
The anode lead wire 96 is connected to the ground plane or zero volts. The cathode 88 is connected to a variable source of between 50 and preferably 2000 volts negative with respect to the grounded anode 94, but may be couple to a voltage of up to several tens of thousands of volts negative with respect to such anode 94. To enhance performance of the invention, the negative voltage may be supplied in the form of pulses having a duty cycle between 0.001 and 0.5
The winding 98 is energized with an alternating voltage such as to provide a current flow of typically between 0.5 and 1.5 amps initially. The frequency of the applied alternating voltage should be variable from DC up to 15 KHz and may, in addition, be synchronous with pulses applied to the cathode 88.
Under these conditions, a plasma arc will strike close to the cathode 88. The intensity and frequency of the current flowing in winding 98 may be adjusted to provide for the removal of the plasma arc from the immediate vicinity of the cathode 88 to avoid excessive evaporation of the material from the cathode 88.
The volume of electrolyte pumped through chamber 80 and past the plasma arc may be varied such as to stabilize the temperature of such electrolyte in a closed system at below its boiling point.
Heat may be extracted from the electrolyte by passing it through a heat exchanger before its re-introduction into the chamber 80. Provision may be made to top-off the water-deuterium content of the electrolyte as this becomes depleted by operation of the apparatus. The system may operate at a range of pressures to facilitate heat removal.
A further apparatus for carrying out the invention, similar to that of Figure 4, is shown in Figure 5 on a scale of approximately 1:2:5. It comprises a borosilicate reaction tube 100 supported at one end on a machined nylon support bridge 102. A second machined nylon element 104 is mounted across the other end of the tube. The bridge 102 and element 104 are clamped against the tube 100 by 8 mm threaded stainless steel studs 110.
A first cathode 106 is in the form of a nickel wire mesh. It is mounted towards one end of tube 100 on a stainless steel support 108. Electrical connection to the first cathode 106 is via a PVS-sleeved wire (not shown).
A second cathode 112 consists of a 0.5mm diameter length of tungsten wire provided within a drilled macor ceramic sheath 114, which is in turn placed within a 10 mm stainless steel tube 116. Tube 116 passes through the support 102 and has a perspex end cap 118 on the external end through which the second cathode 112 passes. A PVC funnel 120 is provided around the second cathode and is tapered towards it, with the cathode tip adjacent the narrower open end thereof. The funnel is supported on sleeves 121 provided on the stainless steel support 108.
The anode comprises a 0.25 mm diameter platinum wire 122 which is connected at one end within the tube 100 to a sheet of platinum foil 124. Like the second cathode 11s, the anode is provided within a 10 mm diameter stainless steel tube 126, which passes through nylon element 104 and is closed at its external end by a Perspex end cap 128. Platinum wire 122 passes through the end cap 128.
A plasma deflection coil 130 is mounted within tube 100 between the anode 124 and cathodes 106, 112. Electrical power is fed to the coil via connectors 132.
Electrolyte is supplied to the tube 100 via a brass inlet 134 provided through the support bridge 102 and flows out through nylon element 104 via a brass outlet 136. An additional brass outlet 138 also is provided in nylon element 104 to allow the electrolyte to be sampled during operation of the apparatus. Fuse holders and cable connectors for the apparatus are provided in a unit 140 mounted on the support bridge 102.
The apparatus of Figure 5 is operated in a similar manner to that of Figure 4, as discussed above. The primary distinction is that two cathodes 106, 112 are employed in place of a single cathode. In use, electrolyte is fed through the tube 100, past the electrodes, from inlet 134 to outlet 136. A pulsed voltage is applied to the first cathode 106 such that a layer of metal hydride is formed on its surface during the voltage pulses and subsequently dissociates to form nascent monoatomic H/D. The applied voltage characteristics are selected to optimize the production rate of the monoatomic H/D. These products are channeled toward the second cathode 112 by the funnel 120. A voltage is applied to the second cathode 112 to generate a plasma discharge thereat.
The characteristics and magnitudes of the voltages applied to the first and second cathodes may be similar, but it may be advantageous for different duty periods to be employed for respective cathodes. This cathode arrangement with the second cathode downstream of the first seeks to maximize contact between the monoatomic H/D and the plasma and therefore the efficiency of the apparatus. This is further assisted by the funnel 120.
Energy Generation
Abstract
Methods and apparatus are described for releasing energy from hydrogen and/or deuterium atoms. An electrolyte is provided which has a catalyst therein suitable for initiating transitions of hydrogen and/or deuterium atoms in the electrolyte to a subground energy state. A plasma discharge is generated in the electrolyte to release energy by fusing the atoms together.
Description
[0001] The present invention relates to the generation of energy, and more particularly to the release of energy as a result of both a state-transition in hydrogen and fusion of light atomic nuclei.
[0002] Normally, fusion processes are able to be initiated only at extremely high temperatures, as found in the vicinity of a nuclear fusion (uranium or plutonium) detonation. This is the principle of most thermonuclear bombs. Such a release of energy is impractical as a means of providing the power to generate electricity and heat for distribution, as it occurs too rapidly with too high a magnitude for it to be manageable.
[0003] In recent years, many attempts have been made to initiate controlled fusion processes at high temperatures by the enclosure of a region of plasma-discharge within a confined space, such as a toroidal chamber, using electromagnetic restraint. Such attempts have met with little commercial success to date as systems which employ such a technique have so far consumed more energy than they have produced and are not continuous processes.
[0004] Another approach which has been attempted in order to achieve fusion of light nuclei has been the so-called "cold fusion" technique, in which deuterium atoms have been induced to tunnel into the crystal lattice of a metal such as palladium during electrolysis. It is claimed that the atoms are forced together in the lattice, overcoming the repulsive electrostatic force. However, no clear and unambiguous demonstration of successful cold fusion has yet been presented publicly.
[0005] The present invention provides a method of releasing energy comprising the steps of providing an electrolyte having a catalyst therein, the catalyst being suitable for initiating transitions of hydrogen and/or deuterium atoms in the electrolyte to a sub-ground energy state, and generating a plasma discharge in the electrolyte. The applicants have determined that this method generates substantially more energy than the power input used to generate the plasma, whilst doing so in a controllable manner.
[0006] Preferably, the plasma discharge is generated by applying a voltage across electrodes in the electrolyte and an intermittent voltage has proved particularly beneficial in increasing the level of energy generation. It also provides a means of controlling the process to maintain a consistent level of energy production over a significant period of time.
[0007] The application of a voltage higher than that necessary to generate plasma is also beneficial to the process and will be typically in the range 50V to 20000V and preferably between 300 and 2000V, but may be higher than 20000V, whereas in conventional electrolysis techniques low voltages of about 3 volts are used and applied continuously across the electrodes.
[0008] The applied voltage may be DC or provided at a switching frequency of up to 100 kHz. The duty cycle of the applied voltage is preferably in the range 0.5 to 0.001, but may be even lower than 0.001. During the pulse period a monomolecular layer of metal hydride may be formed at the cathode-Helmholtz layer interface and subsequently decays to form gas in the nascent state comprising monatomic hydrogen and/or deuterium. The waveform of the applied voltage may be substantially square shaped. Whilst application of DC to the electrode does produce the metal hydride and monatomic hydrogen and/or deuterium, the use of a pulsed voltage has been found to be more efficient as most dissociation of the hydride then occurs between the pulses.
[0009] In applications where the electrolyte is flowed past the electrodes it may be preferable to use two separate cathodes, the first of which will be engineered to optimise production of hydrogen/deuterium atoms and the second of which will provide the plasma discharge. In this instance the direction of flow of the electrolyte is from first to second cathode. The design of the apparatus seeks to direct the flow of electrolyte to maximise contact of monatomic hydrogen or deuterium atoms with the plasma. The characteristics and magnitudes of the voltages applied to each cathode are preferably similar, but may have different duty periods.
[0010] In a preferred embodiment, the cathode design and applied voltage are such as to provide a current density of 400,000 amps per square meter or even greater. More preferably, the current density at the cathode is 500,000 amps per square meter or above.
[0011] In carrying out a preferred method in accordance with the invention, it has been found that the process may be assisted by initial heating of the electrolyte, which may be water or a salt solution, prior to applying electrical input to the vessel. A temperature in the range 40 to 100.degree. C., or more preferably 40 to 80.degree. C., has been found to be particularly beneficial.
[0012] The ratio of water to deuterium oxide (D.sub.2O) in the electrolyte may be varied to control the energy generation. In some circumstances it may be preferable to use "light" water H.sub.2O alone and in others to use D.sub.2O alone. Additionally, the amount of catalyst added to the electrolyte may be varied as a controlling factor and preferably lies in the range 1 to 20 mMol.
[0013] In preferred embodiments, the method includes the step of generating a magnetic field in the region of the electrodes. The intensity and/or frequency of the current used to generate the field may be adjusted to move the plasma discharge away from the electrode from which it is struck in order to minimise erosion and extend the operating life of the system. Only slight separation may be required to achieve this effect.
[0014] In further preferred embodiments, the heat generated by the process may be removed and utilised by way of a number of known and proven technologies including the circulation of the electrolyte through a heat exchanger, or using heat pipes to produce heating, or alternatively to produce electricity using a pressurised steam cycle or a low-boiling-point fluid turbine cycle, or by other means.
[0015] The present invention further provides apparatus for carrying out methods disclosed herein comprising an anode, first and second cathodes, a reaction vessel having an inlet and an outlet, means for feeding an electrolyte through the vessel from its inlet to its outlet, the electrolyte having a catalyst therein suitable for initiating transitions of hydrogen and/or deuterium atoms in the electrolyte to a sub-ground energy state, means for applying a voltage across the anode and the first cathode to form hydrogen and/or deuterium atoms, and means for applying a voltage across the anode and second cathode to generate a plasma discharge in the electrolyte, the second cathode being downstream from the first cathode.
[0016] During the methods described herein, atoms of hydrogen and/or deuterium are believed to undergo a fundamental change in their structure by exchange of photons with salts in solution. The applicants believe that this change, and the observed phenomena, can be explained as set out below.
[0017] It is well known that a system comprising a spherical shell of charge (the electron path) located around an atomic nucleus constitutes a resonant cavity. Resonant systems act as the repository of photon energy of discrete frequencies. The absorbtion of energy by a resonant system excites the system to a higher-energy state. For any spherical resonant cavity, the relationship between a permitted radius and the wavelength of the absorbed photon is:
2.pi.r=n.lambda.
[0018] where n is an integer
[0019] and .lambda. is the wavelength
[0020] For non-radiating or stable states, the relationship between the electron wavelength and the allowed radii is:
2.pi.[nr.sub.1]=2.pi.r.sub.(n)=n.lambda..sub.(1)=.lambda..sub.(n) (2)
[0021] where
[0022] n=1
[0023] or
[0024] n=2, 3, 4 . . .
[0025] or p1 n=1/2, 1/3, 1/4
[0026] and
[0027] .lambda..sub.(1)=the allowed wavelength for n=1
[0028] r.sub.(1)=the allowed radius for n=1
[0029] In a hydrogen atom (and the following applies equally to a deuterium atom), the ground state electron-path radius can be defined as r.sub.(O). This is sometimes referred to as the Bohr radius, a.sub.O. There is normally no spontaneous photon emission from a ground state atom and thus there must be a balance between the centripetal and the electric forces present. Thus:
[m.sub.(e).v.sub.1.sup.2]/r.sub.(O)=Ze.sup.2/(4.pi...epsilon..sub.(O).r.su- b.(O).sup.2) (3)
[0030] where
[0031] m.sub.(e)=electron rest mass
[0032] v.sub.1=ground state electron velocity
[0033] e=elementary charge
[0034] .epsilon..sub.(O)=electric constant (sometimes referred to as the permittivity of free space)
[0035] Z=atomic number (for hydrogen, 1)
[0036] Looking first at the excited (higher energy) states, where the hydrogen atom has absorbed photon(s) of discrete wavelength/frequency (and hence energy), the system is again stable and normally non-radiating, and to maintain force balance, the effective nuclear charge becomes Z.sub.eff=Z/n, and the balance equation becomes:
[m.sub.(e).v.sub.n.sup.2]/nr.sub.(O)=[e.sup.2/n]/(4.pi...epsilon..sub.(O).- [nr.sub.(O)].sup.2) (4)
[0037] where
[0038] n=integer value of excited state (1, 2, 3 . . . )
[0039] v.sub.n=electron velocity in the nth excited state
[0040] The absorbtion of radiation by an atom thus results in an excited state which may decay to ground state, or to a lower excited state, spontaneously, or be triggered to do so, resulting in the re-release of a quantum of energy in the form of a photon. In any system consisting of a large number of atoms, transitions between states are occurring continuously and randomly and this activity gives rise to the observable spectra of emitted radiation from hydrogen.
[0041] Each value of n corresponds to a transition which is permitted to occur when a resonant photon is absorbed by the atom. Integer values of n represent the absorbtion of energy by the atom.
[0042] Fractional values for n are allowed by the relationship between the standing wavelength of the electron and the radius of the electron-path, given by (2), above. To maintain force balance, transitions involving fractional values for n must effectively increase the nuclear charge Z to a figure Z.sub.eff, and reduce the radius of the electron-path accordingly. This is equivalent to the atom emitting a photon of energy while in the accepted ground state, effecting a transition to a sub-ground state. Because the accepted ground state is a very stable one, such transitions are rarely encountered but the applicants have discovered that they can be induced if the atom is in close proximity to another system which acts as a "receptor-site" for the exact energy quantum required to effect the transition.
[0043] The emission of energy by a hydrogen atom in this way is not limited to a single transition "down" from ground state, but can occur repetitively and, possibly, transitions to 1/3, 1/4, 1/5 etc states may occur as a single event if the energy balance of the atom and the catalytic system is favourable. Of course, the usual uncertainty principles forbid the determination of the behaviour of any individual atom, but statistical rules govern the properties of any macroscopic (>10.sup.9 quanta) system.
[0044] When a "ground-state" hydrogen atom emits a photon of around 27 eV, the transition occurs to the a.sub.O/2 state as demonstrated above and the effective nuclear charge increases to +2e. A new equilibrium for the force balance is now established. The electron path radius is reduced. The potential energy of the atom in its reduced radius-state is given by
V=-{Z.sub.(eff)e.sup.2/[4.pi..epsilon..sub.(O)/2)]}=-{4.times.27.178}=-108- .7 eV
[0045] The kinetic energy, T, of the reduced electron path is given by
T=-[V/2]=54.35 eV
[0046] Similarly, it can be seen that the kinetic energy of the ground state electron path is about 13.6 eV. Thus there is a net change in energy of about 41 eV for the transition:
H{Z.sub.(eff)=1; r=a.sub.(O)} to H{Z.sub.(eff)=2; r=a.sub.(O)/2}
[0047] That is to say, of this 41 eV, about 27 eV is emitted as the catalytic transfer of energy occurs, and the remaining 14 eV is emitted on restablisation to the force balance.
[0048] The radial "ground-state" field can be considered as a superposition of Fourier components. If integral Fourier components of energy equal to m.times.27.2 eV are removed, the positive electric field inside the electron path radius increases by
(m).times.1.602.times.10.sup.-19C
[0049] The resultant electric field is a time-harmonic solution of the Laplace equations in spherical co-ordinates. In the case of the reduced radius hydrogen atom, the radius at which force balance and the non-radiative condition are achieved is given by
r.sub.(m)=a.sub.(O)/[m+1]
[0050] where m is an integer.
[0051] From the energy change equations given above, it will be appreciated that, in decaying to this radius from the so-called "ground-state", the atom emits a total energy equal to
[(m+1).sup.2-1.sup.2].times.13.59 eV (5)
[0052] The applicants have found that such energy emissions as take place according to (5), above, only appear to occur when the hydrogen or deuterium is found in the monatomic (or so-called "nascent") state. Molecular hydrogen might be made to behave similarly, but the transition is more difficult to achieve owing to the higher energies involved.
[0053] In order to achieve the transition in monatomic hydrogen (H) or deuterium (D), it is necessary to accumulate the molecular form in the gas phase on a substrate such as nickel or tungsten which favours the dissociation of the molecule. As well as being dissociated into the monatomic form, the hydrogen or deuterium should be bound to the catalytic system to initiate the reaction. The preferred method of achieving this is by electrolysis using cathode material which favours dissociation.
[0054] The applicants have discovered that the catalytic systems which encourage transitions to sub-ground-state energies are those which offer a near-perfect energy couple to the [m.times.27.2] eV needed to "flip" the atom of H or D. It appears from experiment that the effective sink of energy provided by the catalyst need not be precisely equal to that emitted by the atom. Successful transitions have been achieved when there is an error of as much as .+-.2% between the energy emitted by the atom and that absorbed by the catalytic system. One possible explanation for this is that, in a macroscopic sized system, although the transitions are initiated by a close match in energy level, such discrepancies as arise are manifested as an overall loss or gain in the kinetic energies of the recipient ionic systems. It is thought that spectroscopic analysis of active H or D catalytic systems may provide evidence of this.
[0055] One catalyst that has been found to initiate the transition to the a.sub.O/n state is rubidium in the Rb+ ionic species. If a salt of rubidium, such as the carbonate Rb.sub.2CO.sub.3 is dissolved in either water or deuterium oxide (heavy water), a substantial dissociation into Rb.sup.+ and (CO.sub.3).sup.2- ions takes place. If the Rb.sup.+ ions are bound closely to monatomic H or D, the transition to the a.sub.O/n state is encouraged by the removal of a further electron from the rubidium ion, by provision of its second ionisation energy of about 27.28 eV. Thus:
Rb.sup.++H{a.sub.(O)/p}+27.28 eV ->
Rb.sup.2++e.sup.-+H{a.sub.(O)/[p+1]}+{[(p+1).sup.2-p.sup.2].times.13.59}eV
[0056] where p represents an integral number of such transitions for any given H and D atom and by spontaneous re-association:
Rb.sup.2++e.sup.-=Rb.sup.++27.28 eV
[0057] Thus, the rubidium catalyst remains unchanged in the reaction and there is a net yield of energy per transition.
[0058] Other catalytic systems can be used which have ionisation energies approximating to [m.times.27.2]eV, such as titanium in the form of Ti.sup.2+ ions and potassium in the form of K.sup.+ ions.
[0059] The applicants believe that the above explanation is consistent with currently accepted quantum theory as discussed below.
[0060] Commencing with the equations of Rydberg and Schrodinger it can be shown that fractional numbers for the quantum energy states in hydrogen yield possible transitions which result in emissions at frequencies which are in accord with observed UV and X-ray spectra. It is therefore possible that the conditions conducive to initiating such transitions may be artificially reproduced in the laboratory under certain circumstances.
[0061] The Rydberg formula for the frequency of emitted radiation from a transition in monatomic hydrogen is:
v=R.sub.(h)c(1/n.sub.(2).sup.2-1/n.sub.(1).sup.2)
[0062] where:
[0063] v is the frequency of the emitted photon
[0064] R.sub.(h) is Rydberg constant, 1.097373 c 10.sup.7 m.sup.-1
[0065] c is the speed of light in vacuo, 2.997.times.10.sup.3 ms.sup.-1
[0066] and
[0067] n.sub.(1), n.sub.(2) are the transition states
[0068] It can be seen from the above that, if the resultant energy state of the hydrogen atom is that which requires n.sub.(2) to be equal to 1/2, emissions will occur which are of higher frequency than the observed Lyman 2-1 transition in the ultra-violet at 2.467.times.1.degree..sup.15 Hz (about 121 nm). There is, indeed, an observed emission at a wavelength of about 30.8 nm, which appears to be confirmed by recent studies of galactic cluster emissions by Bohringer et al (Scientific American, January 1999) and it is difficult for the inventor to conceive of any other quantum-mechanical event which would give rise to such an emission, other than a transition, in accord with the above theory, from 1 to 1/2 in nascent hydrogen.
[0069] As can be seen from the above use of the standard Rydberg equation, such behaviour of hydrogen in the monatomic state views the conventional hydrogen "ground-state" as one of many stable electronically-preferred states for single H atoms.
[0070] To summarise, a proliferation of H or D atoms is produced which may have had significantly diminished electron-path-radii by virtue of exchange of photons with their environment. These atoms appear to be relatively unreactive chemically and appear not to readily take the molecular form H-H or D-D. This is a fortunate property which has significance and enables fusion pathways, as described below.
[0071] The fusion of light nuclei, hydrogen and deuterium, to form heavier elements such as helium is one which has traditionally been encouraged by subjecting the reactants to extremes of temperature and pressure. This has been necessary because there is a large electric charge barrier to overcome in order to bring nuclei close enough for fusion to occur.
[0072] Using atoms with diminished electron path radius, adjacent nuclei may experience a corresponding reduction in electric barrier and internuclear separations may become smaller. With reductions in internuclear separation, fusion processes become more probable, and more easily occasioned.
[0073] There are two principle fusion pathways for deuterium atoms. The first is:
.sup.2.sub.1D+.sup.2.sub.1D=.sup.3.sub.2He+.sup.1.sub.0n
[0074] where two deuterium nuclei fuse to produce an isotope of helium and a free neutron, which subsequently decays (half-life 6.48.times.10.sup.2S), with emission of a .beta..sup.- particle of medium energy (about 0.8 Mev), and a type of neutrino, to become a stable proton.
[0075] The second is:
.sup.2.sub.1D+.sup.2.sub.1D=.sup.3.sub.1T+.sup.1.sub.1H
[0076] where the two deuterium nuclei fuse to produce the isotope of hydrogen known as tritium (T) and a free stable proton. The tritium eventually decays (half-life 12.3 years), with emission of a .beta..sup.- particle of very low energy (about 0.018 MeV), to become .sup.3.sub.2He
[0077] Of the two, the second fusion path is preferred for the peaceful exploitation of its energy yield, because the fusion products are (relatively) harmless on production, and decay to completely innocuous species within a short time, emitting radiation which can be effectively shielded by a thin sheet of kitchen foil or by 10 mm of acrylic plastic, for example.
[0078] When deuterium nuclei are forced together under high temperature and pressure conditions (as in a thermonuclear bomb), there is a greater than 50% probability for the first pathway to be the dominant one. This is because the high temperature process takes no account of nuclear alignment at the point of fusion. It is actually a matter of forcing nucleic together indiscriminately and hoping that enough fuse to produce an explosion. However, the applicants believe, in accord with established theory, that it is the alignment of the nuclei with respect to the charges in each nucleus which ultimately determines the favourable fusion path.
[0079] In order to achieve a higher probability for the second, less hazardous, pathway, the approaching nuclei need to have time to align electrostatically such that the proton-proton separation is at a maximum. This can only be achieved at far lower energies than those found in a thermonuclear bomb. By the use of entities with diminished electron-path-radii, and correspondingly potentially smaller internuclear distances, fusion can be initiated at lower temperatures (and consequently lower energies), allowing for the charge-related alignment necessary to achieve a high probability for the second, tritium-forming, pathway. By introducing deuterium of diminished electron-path-radius into a plasma discharge which is confined within the water in the vessel itself, fusion is may be initiated. Temperatures of the order of 6000 K are obtained within certain plasma discharges and this, coupled with multiple quantum transitions to produce deuterium of diminished electron-path-radius, produces a substantial yield of energy from the two-stage process.
[0080] Another possible but less likely fusion pathway for hydrogen atoms is:
.sup.1.sub.1H+.sup.1.sub.1H=.sup.2.sub.1D+.beta..sup.++.tau.
[0081] whereby .beta..sup.+ is produced as one of the products.
[0082] Embodiments of the invention will now be described by way of example and with reference to the accompanying schematic drawings, wherein:
[0083] FIG. 1 shows an apparatus for carrying out a method in accordance with the invention on a relatively small scale;
[0084] FIG. 2 shows a system for operating and measuring the performance of the apparatus of FIG. 1;
[0085] FIG. 3 shows a circuit diagram high voltage, high frequency switching circuit for the system of FIG. 2;
[0086] FIG. 4 shows an apparatus for carrying out a method in accordance with the invention on a larger scale than that of the FIG. 1 apparatus; and
[0087] FIG. 5 shows a further apparatus for carrying out a method of the invention which includes two cathodes.
[0088] The apparatus of FIG. 1 enables the generation of energy according to the principles of the invention in the laboratory. Any risk of thermal runaway is minimised, whilst demonstrating that the level of energy release from the two stages is far in excess of that which would result from any purely chemical or electrochemical activity. It also enables easy calorimetry, safe ducting away of off-gases, and of subsequent extraction of liquid for titration (to demonstrate that no chemical action takes place during the operation of the apparatus).
[0089] A 250 ml beaker is provided with a glass quilt or expanded polystyrene surround 6 to act as insulation. This can include an inspection cut-out so that the area around the cathode 9 can be observed from outside. The beaker contains 200 ml of water, into which is dissolved a small quantity of potassium carbonate so as to give a solution of approximately 2 mMol strength. A platinum lead wire 1 is earthed to the laboratory reference ground plane. The anode 10, a sheet of platinum foil of approximately 10 mm.sup.2 in area, is attached to this lead wire by mechanical crimping. A digital thermometer 2 is inserted into the liquid in the vessel. A 0.25 mm diameter tungsten wire cathode 9 is sheathed in borosilicate glass or ceramic tube 4 and sealed at the end immersed in the electrolyte so as to expose 10 mm to 20 mm of wire in contact with the liquid. The entire assembly of lead wires and the thermometer is carried by an acrylic plate 5 which enables of easy dismantling and inspection of the apparatus.
[0090] A supply of up to 360 volts DC, capable of supplying up to 2 amperes, is arranged external to the described apparatus. The positive terminal of this supply is connected to the laboratory reference ground plane and the negative terminal is connected to one pole of an isolated high-voltage switching unit. The other pole of the switch is connected to the tungsten wire cathode 9 externally of the apparatus.
[0091] To operate the apparatus, the solution 8 is initially brought up to between 40.degree. C. and 80.degree. C. either by preheating outside the apparatus or by passing power through a heating element in the solution (not shown). When the solution is between these temperatures it is either transferred to the above apparatus or, if a heating element is used, this is turned off.
[0092] With all connections made as described, the switch is set to operate at a duty cycle of 1% and a pulse repetition frequency of 100 Hz. It will be seen through the inspection cut-out that an intense plasma-arc is intermittently struck under the water at or near the cathode. If equipment is available to monitor the current drawn, it will be seen that the system consumes in the region of 1 watt when the switching circuits is operating. It will be seen by the rapid rise in temperature in the apparatus that far more energy is being released than can be accounted for by the electrical input. As a comparison, a heater element can be substituted for the electrodes and operated 1 watt and the effects observed. There is really no need for sophisticated calorimetry to verify that large quantities of energy are being released close to the cathode of the equipment, such is the magnitude of the reaction for the process, as compared to a test with a resistive heating element of the same input power.
[0093] The data obtained from a representative one-hour session with this apparatus as shown as Table 1, below:
1 Pre Run Measurements Commencing volume of electrolyte 0.200 l Commencing temperature of cell 39.200.degree. C. Laboratory ambient temperature 20.500.degree. C. Spec. heat capacity of vessel 70.300 J .multidot. .degree. C..sup.-1 Spec. heat capacity of electrolyte 4180.000 J .multidot. I.sup.-1 .multidot. .degree. C..sup.-1 Steady RMS voltage 4.000 volts Steady RMS current 0.067 Amps Post Run Results Duration of input 3600.000 secs Final volume of electrolyte 0.180 l Final temperature of cell 93.600.degree. C. Steady RMS voltage 6.700 volts Steady RMS current 0.122 Amps Time-averaged power in 0.506 watts Results Summary Vessel Gain 3824.320 Joules Electrolyte gain 43181.740 Joules Radiated power 38681.030 Joules Evaporated loss 48509.240 Joules TOTAL ENERGY IN 1820.070 Joules TOTAL ENERGY OUTPUT 134196.300 Joules
[0094] It can be seen from this table that the total energy input during this test was measured at 1820 Joules and, taking as a rough guideline that 200 ml of water requires the input of 838 joules of energy to raise it by 1.degree. C., then by direct heating the water would be expect to rise by some 2.degree. C., bearing in mind radiative losses. In fact, during the experiment the water temperature was raised from 39.2.degree. C. to 93.6.degree. C. and considerable steam was also liberated. Furthermore, the calculated energy output of 134196 Joules does not take account of secondary effects such as light-energy output and Faradaic electrolysis.
[0095] A system suitable for operating the apparatus of FIG. 1 is illustrated in a block diagram in FIG. 2. A pulse generator 20 supplies a variable duty-cycle pulse waveform to a high voltage switch unit 22. The pulse waveform may be monitored on an oscilloscope 24 and its repetition frequency is displayed on a first frequency counter 26. A second frequency counter 28 is provided to monitor the clock speed of the switch unit 22. Power supply 30 is operable to apply a voltage between 0 and 360 V to an electrode of the apparatus 12, shown in FIG. 1. The voltage level may be read from a digital multimeter 32. The RMS voltage across the electrodes 9 and 10 is indicated on a multimeter 34 and the RMS current passing between the electrodes is shown on another multimeter 36, by measuring the voltages developed across a 1 ohm resistor 37. The temperature in the apparatus 12 is indicated on a dip temperature probe 38. The switch unit 22 may be bypassed by a push button switch 39 to apply a constant voltage across the electrodes.
[0096] A circuit diagram of the switch unit 22 is shown in FIG. 3. In the system of FIG. 2, input 40 is connected to the output of pulse generator 20. The output 42 of the switch unit is connected to the cathode of the apparatus 12. Two NAND gates 44 and 46 are two fourths of a Schmitt-trigger 2 input NAND gate chip type 4093. NAND gate 44 operates as an astable multivibrator, with its repetition frequency set by a preset resistor 45. The output of gate 44 is fed to one input of NAND gate 46, the other input forming circuit input 40. The output of NAND gate 46 is connected to a three transistor amplifier consisting of transistors 48, 50 and 52. The amplifier is in turn connected to one end of the primary of a transformer 54, the other end being connected to earth. The transformer output is fed to a bridge rectifier formed from diodes 56, 58, 60 and 62.
[0097] The rectifier output is fed via a resistor 64 to the gate of an insulated gate bipolar transistor 66 (IGBT). The load of the apparatus 12 is connected in the drain circuit of the IGBT. A 15 kV diode 68 is connected between the drain and the source of the IGBT 66 to protect the IGBT from the sizeable EMI emissions from plasma discharges in the apparatus 12 and avoids damage to this sensitive semiconductor. A further diode 70 is provided between the drain of the IGBT and the circuit output 42 to act as an EMI blocker in a similar way. A standard 20 mm 5A quick-blow fuse 69 is connected between the source of the IGBT and ground in order to protect the device against overcurrent.
[0098] The operation of the circuit of FIG. 3 is as follows. The repetition frequency is NAND gate 44 is preferably set to between 4 and 6 MHz. Pulse generator 20 is adjusted to set the duty of the switching. On receipt of an external pulse from the generator, NAND gate 46 passes a packet of 4 to 6 MHz square waves to the amplifier. The amplifier has considerable current gain and enables the primary of the transformer 54 to be driven resonantly with the RC circuit formed by capacitor 72 and resistor 74 which are connected in parallel therewith. The transformer 54 has a step-up ratio of 2:1 and a 4 to 6 MHz signal of approximately 19 volts appears across the bridge rectifier. The impedance of the rectifier output is essentially determined by a parallel resistor 76, such that the switch-on and switch-off time of the IGBT 66 is very fast. Thus, there is never a point in the operation of the device when it is dissipating any measurable power. The load of the apparatus 12 is placed in the drain circuit of the IGBT, which is therefore operating in "common-source" made to ensure that its source terminal never rises above high-side ground potential. This, again, is a configuration which uses excess input power. This circuit ensures a rise time of the switched waveform which is less than 10 nS and a fall time which can be as low as 30 nS at modest supply voltages.
[0099] Preferred component values and types for the circuit of FIG. 3 are as follows:
[0100] Transistor 4, 50--2N 3649
[0101] Transistor 52--2N 3645
[0102] Diodes 56, 58, 60, 62--BAT85 Schottky
[0103] Transformer 54--RS195-460
[0104] IGBT 66--GT8Q101
[0105] Diode 68--15 kv EHT
[0106] Diode 70--1N1198A
2 Resistor Value (.OMEGA.) Capacitor Value 47 1.8k 49 10 pF 51 33 55 33 nF 53 220 72 22 pF 74 56 76 560 64 56
[0107] A second apparatus for carrying out the invention is illustrated in FIG. 4. This apparatus comprises a tubular chamber 80, which may be constructed from a nonmagnetic metal or metal alloy material such as, but not exclusively, aluminium or Duralumin, or may alternatively be constructed from a non-permeable ceramic material or from borosilicate glass. The tubular chamber 80 is constructed in flanged form to allow of its incorporation into a system of pipework via flanges 82 and 84 and gaskets 86. Entering the chamber 80 are two electrodes, the cathode 88 being sheathed in an insulating glass or ceramic tube 90 and shaped so as to present itself along the axis of the chamber 92. The anode 94 is connected to a similar insulated wire 96 and is shaped so as to present a circular plate opposite the cathode 88. The distance between the cathode tip and the anode plate should be approximately equal to the radius of the chamber 80. The cathode may be constructed from tungsten, zirconium, stainless steel, nickel or tantalum, or any other metallic or conductive ceramic material which may contribute to, or occasion, the dissociative process described above. The anode may be constructed from platinum, palladium, rhodium or any other inert material which does not undergo any significant level of chemical interaction with the electrolyte.
[0108] Surrounding the chamber 80, and concentric with it, is a winding 98 of enamelled copper or silver wire of diameter 0.1 to 0.8 mm consisting of up to several thousand turns of the wire. The purpose of this winding 98 is to create an axial magnetic field inside the chamber 80.
[0109] Electrolyte comprising deuterium oxide, in combination with ordinary "light" water in varying proportions, and containing high-molarity salts of, but not exclusively of, potassium, rubidium or lithium, or combinations of such salts, is pumped through the chamber 80, in a direction such that the anode is downstream of the cathode.
[0110] The anode lead wire 96 is connected to the ground plane or zero volts. The cathode 88 is connected to a variable source of between 50 and preferably 2000 volts negative with respect to the grounded anode 94, but may be coupled to a voltage of up to several tens of thousands of volts negative with respect to such anode 94. To enhance performance of the invention, the negative voltage may be supplied in the form of pulses having a duty cycle between 0.001 and 0.5.
[0111] The winding 98 is energised with an alternating voltage such as to provide a current flow of typically between 0.5 and 1.5 amps initially. The frequency of the applied alternating voltage should be variable from DC up to 15 kHz and may, in addition, be synchronous with pulses applied to the cathode 88.
[0112] Under these conditions, a plasma arc will strike close to the cathode 88. The intensity and frequency of the current flowing in winding 98 may be adjusted to provide for the removal of the plasma arc from the immediate vicinity of the cathode 88 to avoid excessive evaporation of the material from the cathode 88.
[0113] The volume of electrolyte pumped through chamber 80 and past the plasma arc may be varied such as to stabilise the temperature of such electrolyte in a closed system at below at its boiling point.
[0114] Heat may be extracted from the electrolyte by passing it through a heat exchanger before its re-introduction into the chamber 80. Provision may be made to top-up the water/deuterium content of the electrolyte as this becomes depleted by operation of the apparatus. The system may operate at a range of pressures to facilitate heat removal.
[0115] A further apparatus for carrying out the invention, similar to that of FIG. 4, is shown in FIG. 5 on a scale of approximately 1:2.5. It comprises a borosilcate reaction tube 100 supported at one end on a machined nylon support bridge 102. A second machined nylon element 104 is mounted across the other end of the tube. The bridge 102 and element 104 are clamped against the tube 100 by 8 mm threaded stainless steel studs 110.
[0116] A first cathode 106 is in the form of a nickel wire mesh. It is mounted towards one end of tube 100 on a stainless steel support 108. Electrical connection to the first cathode 106 is via a PVC-sleeved wire (not shown).
[0117] A second cathode 112 consists of an 0.5 mm diameter length of tungsten wire provided within a drilled macor ceramic sheath 114, which is in turn placed within a 10 mm stainless steel tube 116. Tube 116 passes through the support 102 and has a perspex end cap 118 on the external end through which the second cathode 112 passes. A PVC funnel 120 is provided around the-second cathode and is tapered towards it, with the cathode tip adjacent the narrower open end thereof. The funnel is supported on sleeves 121 provided on the stainless steel support 108.
[0118] The anode comprises an 0.25 mm diameter platinum wire 122 which is connected at one end within the tube 100 to a sheet of platinum foil 124. Like the second cathode 112, the anode is provided within a 10 mm diameter stainless steel tube 126, which passes through nylon element 104 and is closed at its external end by a perspex end cap 128. Platinum wire 122 passes through the end cap 128.
[0119] A plasma deflection coil 130 is mounted within tube 100 between the anode 124 and cathodes 106, 112. Electrical power is fed to the coil via connectors 132.
[0120] Electrolyte is supplied to the tube 100 via a brass inlet 134 provided through the support bridge 102 and flows out through nylon element 104 via a brass outlet 136. An additional brass outlet 138 is also provided in nylon element 104 to allow the electrolyte to be sampled during operation of the apparatus. Fuse holders and cable connectors for the apparatus are provided in a unit 140 mounted on the support bridge 102.
[0121] The apparatus of FIG. 5 is operated in a similar manner to that of FIG. 4, as discussed above. The primary distinction is that two cathodes 106, 112 are employed in place of a single cathode. In use, electrolyte is fed through the tube 100, past the electrodes, from inlet 134 to outlet 136. A pulsed voltage is applied to the first cathode 106 such that a layer of metal hydride is formed on it surface during the voltage pulses and subsequently dissociates to form nascent monatomic hydrogen/deuterium. The applied voltage characteristics are selected to optimise the production rate of the monatomic hydrogen/deuterium. These products are channelled towards the second cathode 112 by the funnel 120. A voltage is applied to the second cathode 112 to generate a plasma discharge thereat.
[0122] The characteristics and magnitudes of the voltages applied to the first and second cathodes may be similar, but it may be advantageous for different duty periods to be employed for respective cathodes. This cathode arrangement with the second cathode downstream of the first seeks to maximise contact between the monatomic hydrogen/deuterium and the plasma and therefore the efficiency of the apparatus. This is further assisted by the funnel 120.
US6,290,836
Electrodes
Abstract
An electrode (1) having an active surface for contacting an electrolyte. The electrode (1) comprises first and second metallic materials (2, 3) arranged to provide a number of first metallic material to second metallic material interfaces at the active surface. The invention also relates to a method of making such an electrode (1) and to an electrolysis cell provided with such an electrode (1).
Description
TECHNICAL FIELD
This invention relates to an electrode and to a method of making such an electrode. The invention also relates to a cell incorporating such an electrode as its cathode and to a method of obtaining release of gaseous products from such a cell.
BACKGROUND ART
During electrolysis, the mass of a substance liberated by the passage of an electric current is strictly determined by Faraday's Laws of Electrochemical Deposition. These laws state that:
1. "The amount of chemical change occasioned by the passage of an electric current is proportional to the quantity of electricity passed"; and
2. "The masses of different substances liberated by a given quantity of electricity are proportional to their chemical equivalent weights."
The chemical equivalent weight of any substance is easily determined and remains a fixed standard for that substance under all conditions of electrolytic action. It is usually quoted in m.g.C.sup.-1, 1 Coulomb (C) being the quantity of electricity used when a current of one ampere is passed for one second.
If the chemical equivalent weight is represented by z, the mass, m, of any substance liberated during an electrolytic process is given by:
where I is the current passed in amperes and t is the time in seconds.
During normal electrolytic processes, it is not possible to induce a current to flow through the electrolyte unless the voltage across the electrodes of the electrolytic cell is raised to some specific value, which varies according to the electrolyte and the electrode composition. This voltage, V.sub.d, is known as the Decomposition Voltage. Hitherto, it has not been possible to arrange for electrolytic cells to function at voltages sufficiently low to enable of very low-power inputs to the cell.
Any process which can be arranged to run in such a way that, when the calorific value of a liberated gas is higher than the power required to run the electrolytic process which liberates that gas, will act as a net provider of energy. The apparent surplus of energy coming, in this instance, from the bond dissociation energies of the ions involved in the process.
An example of the operation of an electrolytic cell will serve to illustrate the above points more clearly.
Let us first consider a cell which liberates hydrogen gas by the electrolysis of water containing a standard electrolyte such as H.sub.2 SO.sub.4 or Li.sub.2 SO.sub.4. If such a cell is run such that its terminal voltage is 5 volts and the current being passed through it is 2 amperes, it will require a power source of at least 10 watts, allowing for small losses in wires and contact resistances. The mass, and hence calorific value, of the hydrogen liberated from such a cell will be in accordance with Faraday's Laws and will be proportional to the product of current and time as outlined above. However, the product of current and time is not the same thing as the product of current and voltage, which gave us the power consumption of the cell. In the case of this cell, the power input is given simply by:
P.sub.in =V.times.I
where V is the cell voltage and I is the cell current.
To calculate the power output of such a cell, we need to know how much energy is available from a given mass of hydrogen gas when it combines with oxygen during combustion. This figure is 285 KJ.mol.sup.-1, where 1 KJ (kilojoule) is the energy converted when 1 kilowatt of power is used for a duration of 1 second. Since the chemical equivalent weight of hydrogen is known to be 0.01045 mgC.sup.-1, it can be calculated, according to (1) above, that the cell will yield a mass, m, of hydrogen gas given by
1 mol of hydrogen gas, as molecular hydrogen H.sub.2, has a mass of 2.016 g. Utilising the energy content of hydrogen as it undergoes combustion, we therefore have an energy yield from the cell of: ##EQU1##
It can be seen, therefore, that this conventional cell only produces just over a quarter as much energy from the full combustion of its hydrogen yield as the electrical energy required to make it run. Such a device is not an efficient converter of energy.
Consider now the performance of the same cell if its current of 2 amperes were to flow using a very much smaller potential of only 0.5 volts. The input power is given by the same equation (2) above, namely:
=0.5.times.2=1 W
The output power, however, remains the same as in the 5 volt example, it being dependent solely upon the parameters of current and time.
The 0.5 volt cell, therefore, yields a supply of hydrogen gas which is capable of being burned to provide some 2.9 times the electrical energy input to the cell.
In the past it has not been possible to cause electrolysis cells to operate at the small voltages necessary to achieve this kind of "energy multiplier" effect. The natural barrier of the established decomposition voltage always halted the process some way before the over-unity effects of the cell became evident.
DISCLOSURE OF THE INVENTION
The present invention seeks to provide an electrode which when used in an electrolytic cell enables current to pass at a low voltage compared with conventional cells. It is also an aim of the invention to enable the generation of a gaseous product form an electrolyte.
According to one aspect of the present invention an electrode having an active surface for contacting an electrolyte, is characterised in that the electrode comprises first and second metallic materials arranged to provide at least one first metallic material to second metallic material interface at said active surface.
Preferably there are a plurality of such interfaces.
Preferably the first metallic material comprises a substrate e.g. of steel, of the electrode and the second metallic material, e.g. nickel or a matrix of nickel and chromium, is plated over regions of the substrate.
According to another aspect of the present invention there is provided an electrolysis cell for obtaining the release of gaseous products by electrolysis, comprising an electrolyte, an anode and a cathode in the form of an electrode according to said one aspect of the present invention. In use of the cell, the current can be passed in such a way that decomposition occurs at a fraction of the usual required voltage. Typically "energy multiplier" effects of the order of 6:1 are achievable.
Suitably the electrolyte comprises dilute sulphuric acid or an aqueous solution of lithium sulphate monohydrate, nickel sulphate hexahydrate, chromium sulphate or palladous chloride.
According to a still further aspect of the invention there is provided a method of making an electrode according to said one aspect of the invention, comprising plating a substrate of a first metallic material with a second metallic material and removing regions of the plated second metallic material to create said active surface with said plurality of first metallic material to second metallic material interfaces.
According to a yet further aspect of the present invention, a method of obtaining release of gas from an electrolysis cell according to said further aspect of the invention, comprises applying a decomposition voltage of no more than 1 volt, preferably no more than 0.8 volts, e.g. from 0.2 to 0.6 volts, across the anode and cathode of the electrolysis cell.
BRIEF DESCRIPTION OF DRAWINGS
An embodiment of the invention will now be described, by way of example only, with particular reference to the accompanying drawing, in which FIGS. 1 to 3 show three stages in the manufacture of an electrode according to the present invention.
BEST MODE FOR CARRYING OUT THE INVENTION
A known electrolyte cell comprises an anode and a cathode as electrodes in an aqueous solution of an electrolyte. If a sufficiently large voltage, i.e. the "emf" of the cell, is applied across the electrodes, gaseous products (hydrogen and oxygen) are released at the electrodes. For any given electrolyte in water, this value lie between 1.250 volts and 2.000 volts, depending upon the ambient conditions in the cell (temperature, electrode metals, degree of wetting, pH of the electrolyte etc.), and is known as the Decomposition Voltage or DV. It is made up of three component voltages, which add arithmetically to give the overall DV for the cell, namely: the hydrogen over-voltage at the cathode; the oxygen over-voltage at the anode; and the electrolyte breakdown voltage.
An electrolytic cell in accordance with the invention differs from known electrolytic cells in that it functions as a so-called Sub-Decomposition-Voltage (hereafter referred to as "SDV") cell which is able to operate at voltages well below the predicted emfs which would be expected by summing the three component voltages above for any given set of cell characteristics.
There are two principal parameters of an SDV electrolytic cell which cause it to function in the way it does. The first parameter is the nature of the electrolyte, and the second (more important) is the physical characteristic of the cathodic electrode. These two parameters are considered below.
Electrolyte
In common with nearly all electrolytic mechanisms, an SDV cell will not work using pure water or even, to any great degree, tap water as the electrolyte. The activity of electrolysis depends upon the migration of ions towards charged surfaces, where they act as either donors or recipients of electrons, and there are simply not enough dissociated ions in pure water to enable this to take place effectively. An electrolyte, as well as dissociating into ions itself, will facilitate to a greater or lesser degree the dissociation of the water in which it is placed. The electolyte material is, nonetheless, recycled and wholly conserved in the process and, once charged, an SDV cell, in common with most other electrolysis devices, requires only to be topped up with water, not fresh electrolyte. Examples of electrolytes which have been successfully employed in SDV cells include dilute H.sub.2 SO.sub.4, lithium sulphate monohydrate, nickel sulphate hexahydrate, chromium sulphate, and palladous chloride, although this is by no means an exhaustive list of the possible substances. Those which function by the release of SO.sub.4.sup.2- ions in solution seem also to perform better when acidified slightly.
The Nature of the Cathode
The cathode of the SDV cell has an active surface comprising two different metallic materials with a plurality of interfaces between the different metallic materials. Conveniently the SDV cathode 1 (see FIG. 3) consists of a substrate 2 of a first metallic material and a plurality of isolated plated region 3 on the substrate 2. Suitably the plated second metallic material comprises nickel, or a matrix of nickel and chromium, so as to create interfaces between the substrate and the plating.
At these interfaces in use of the SDV cell, a number of complex electrochemical interactions take place. When a small voltage is applied across the anode and cathode, H.sub.3 O+ (and other+ve) ions are attracted towards the cathode. These ions are absorbed into the crystal matrix of the nickel plated areas but not into the areas of untreated steel. The sorption process takes place in three main steps, namely: the surface adsorption of the ions, accompanied by their partial dissociation into monatomic hydrogen and water; followed by intergranular rift diffusion of individual atoms of hydrogen between the nickel crystals; and, lastly, lattice diffusion of the same hydrogen atoms from the rifts into the actual lattice of the crystal structure. This is not a clathrate process, there being an immediate association of monatomic H into molecular H.sub.2 within the lattice, accompanied by an increase in pressure. The rate-controlling process is probably the surface adsorption as increased working pressure within the cell appears to have little effect on the rate of hydrogen take-up.
Lattice diffusion continues until the interface between nickel and steel is encountered and it is at this point that molecular hydrogen is released into the adjacent electrolyte. The entire process maintains an equilibrium with the ion-product of the water in the electrolyte, new H.sub.3 O+ and other ions being formed at the same rate as molecular hydrogen is being discharged from the cell. It is thought that there are two catalytic, facilitating, reactions at work. Firstly, the transition from integranular rift diffusion to lattice diffusion is believed to be facilitated by the somewhat unbalanced nature of the two outermost quantum groups in the nickel atom, monatomic hydrogen being "ushered", as it were, by the weak forces within the lattice itself. (Although nascent hydrogen is not itself a polar entity, the existence within any mass of H of two species, ortho- and para-, dependant on Pauli m.sub.s values of + or -1/2, does not rule out some kind of interaction when such a monatomic gas is confined within an electrostatically active crystalline complex.) Secondly, at the small iron-nickel interfaces which occur when the cathode is machined, there is a degree of electron-sharing between adjacent iron and nickel atoms at the periphery of the crystal structure which in some way mitigates in favour of molecular H.sub.2. There are also grounds for considering the existence of free protons within such a intercrystalline confinement and there is nothing in the electrochemistry which would rule this out.
The Anode Process
The anode process differs from that of a conventional cell in that the oxygen over-voltage is rarely exceeded and the reaction at the anode is one of the formation of a (conductive) layer of a matrix of ferrous- and feroso-ferrous-oxide over the plain steel electrode. There is some liberation, albeit slowly, of gaseous oxygen at the anode but this is small in comparison with the ejection of H.sub.2 from the cathode, which occurs prolifically and often (as would be expected given the pressure within the crystalline absorption mechanism at work) with some minor violence when observed under the microscope.
There is, obviously, some likely benefit in obtaining hydrogen from such a process which is relatively free of associated oxygen but, to date, the gaseous mix from experimental SDV cells has not been such as to bring the O.sub.2 level down below the LEL for hydrogen/oxygen mixture, and such cells should not be regarded as being intrinsically safer than conventional ones.
One method of creating an SDV electrode is described below.
The electrode which is to become the cathode in an SDV cell is made by taking a sheet of ordinary mild steel as the substrate 2 and creating on its surface a series of irregularities, in the form of trough regions 4 and raised regions 5 (see FIG. 1), by etching the steel in a bath of concentrated (50-55%) sulphuric acid. The natural impurity of most commonly available mild steel ensures that etching will take place in a random and irregular manner. Mostly, this is caused by the presence of finely divided granular alpha-ferrite which appears to be preferentially attacked by the acid.
After inspection of the surface and the determination of the average size of the nodes or raised regions on the roughened steel (optimally these should be at 0.03-0.05 mm distribution), the surface is passivated in concentrated nitric acid and further passivated in a chromic acid bath.
The roughened surface of the steel substrate 2 is then given a 25-micron coating 6 of nickel by the "electroless" process, also known as auto-catalytic chemical deposition (see FIG. 2). This plating process provides accretion of deposited nickel in the trough regions 4 and thinner deposits of nickel on the raised regions 5.
After coating, the electrode is machined or ground, e.g. using a linishing sander and 120 grit silicon carbide paper belt, to remove the "peaks" of the plated raised regions 5 and in particular to remove the plated nickel from these "peaks" so as to expose the steel of the substrate 2 (see FIG. 3). In this way a plurality of metal-to-metal interfaces are created on the active surface of the cathode between the nickel plated regions on the trough regions 4 of the substrate 2 and the exposed steel surfaces of the substrate. Constant microscopic inspection is required to determine the existence of the correct bi-metallic interfaces on the active surface of the electrode. If the electrode is to be used with only one active surface (SAS electrode), no treatment is given to the other plated surface, which will remain electrochemically inactive during the operation of the cell. If both surfaces are required to work electrolytically (DAS), a similar treatment is given to the other side. After cleaning the electrode in methyl ethyl ketone to remove grease and other machining deposits, it is left immersed in a 0.5N aqueous solution of nickel sulphate hexahydrate at 55.degree. C. for 24 hours, which process acts as an "initiator" for the later complex sequence of ion exchange operations in the active cell.
The present invention envisages a novel cathode and SDV electrolytic cell provided with such a cathode. The invention also teaches a novel method of making such a cathode and a novel method of releasing gaseous products from an SDV cell.
The invention discloses the provision of bi-metallic interfaces on the active, electrolyte-contacting surface of an electrode which produces hitherto unobserved electrochemical phenomena. The use of dissimilar metallic materials on the active surface facilitates lattice diffusion of gases within the crystal structure of the electrode.
An SDV cell according to the invention acts as an "over-unity" cell in respect of hydrogen gas production from the cell. The cell operates at low voltages of no more than 1 volt, preferably no more than 0.8 volt and typically from 0.2 to 0.6 volts. However even lower operating voltages are feasible.

JL Naudin
Laboratory:
http://jlnlabs.imars.com/cfr/html/cfr30.htm
Video of Test Run # 1:
http://jlnlabs.imars.com/cfr/videos/cfrv30a.rm
"The Enhanced Cold Fusion Reactor v3.0"
Description
The Cold Fusion Reactor ( CFR ) v3.0 uses a new enhanced design with a 1000 mL Dewar vessel filled with a 600 mL of demineralized water and 41.5 g of Potassium Carbonate, the electrolyte solution used is 0.5 molar ( 0.5 M, ). The Dewar vessel, used as the container for the CFR v3.0, keeps the strong heat and the light energy produced by the reactor. The reduced output of the Dewar neck avoid some eventual projections of the water outside the vessel.
The CFR v3.0 runs at a very stable regime and the power efficiency measured during all the tests conducted is more than 200%.
The Cathode used is a pure tungsten rod ( W ) 3 mm diameter and 25 mm length from tungsten TIG electrodes (WP) commonly used for TIG and Plasma welding. The Anode used is composed of stainless steel grid maintained with a stainless steel shaft. All the wires connections are made with a 1.5 mm2 copper flexible wire gained with silicon.
The CFR v3.0 is powered with a DC voltage through a bridge rectifier connected through an adjustable isolation transformer to the 220V AC power grid line. The voltage input has been measured with a digital oscilloscope Fluke 123 with a Shielded Test Lead STL 120 ( 1:1, 1 Mohms/225 pF ). The current input has been measured with a current clamp CIE Model CA-60A ( Accuracy DC Amps ±1.5%, AC Amps ±2% (40 Hz - 2 kHz), AC Amps ±4% (2 kHz -10 kHz), AC Amps ±6% (10 kHz - 20kHz) ). The temperature has been measured with a type "K" temp probe ( NiCrNi ) connected on a VC506 digital multimeter ( -20°C to +1200 °C with an accuracy of +/- 3% ).
Run # 1 ~ Test procedure :
1) The temperature
of the K2CO3 solution in the CFR has
been set initially to 83°C.
2) The weight of the CFR has been measured initially, it was
1336 g.
3) The power supply has been switched on continuously and the
Voltage/Current datas has been recorded in the Fluke 123
digital oscilloscope used as a data logger, up to a
temperature of 102°C.
4) Then the weight of the CFR has been measured, it was 1292
g.
The run time of the CFR has been 124.8 seconds.
The Voltage/Current datas logged give an average electrical power input of 574.8 Watts during 124.8 seconds, so this gives :
ELECTRICAL ENERGY INPUT = 71739 Joules
The evaporated water in the CFR during the full boiling was 44 mL. We know that we need 2260 J/g to vaporize water. The temperature rise of the 600 mL was 19°C. So, this gives :
ENERGY OUTPUT = ( 44 x 2260 ) + ( 600 x 19 x 4.18 ) = 147092 Joules
Power OUTPUT =
1178.6 Watts, Net Power Gain = 603.8 Watts
Energy OUTPUT/INPUT = 147092 / 71739 = 2.05
Run # 2 ~ Test procedure :
1) The temperature
of the K2CO3 solution in the CFR has
been set initially to 86°C.
2) The weight of the CFR has been measured initially, it was
1334 g.
3) The power supply has been switched on continuously and the
Voltage/Current datas has been recorded in the Fluke 123
digital oscilloscope used as a data logger, up to a
temperature of 103°C.
4) Then the weight of the CFR has been measured, it was 1286
g.
The run time of the CFR has been 127.2 seconds.
The Voltage/Current datas logged give an average electrical power input of 518.2 Watts during 127.2 seconds, so this gives :
ELECTRICAL ENERGY INPUT = 65918 Joules
The evaporated water in the CFR during the full boiling was 48 mL. We know that we need 2260 J/g to vaporize water. The temperature rise of the 600 mL was 17°C. So, this gives :
ENERGY OUTPUT = ( 48 x 2260 ) + ( 600 x 17 x 4.18 ) = 151116 Joules
Power OUTPUT = 1188
Watts, Net Power Gain = 669.8 Watts
Energy OUTPUT/INPUT = 151116 / 65918 = 2.29
After a lot of tests runs, some small longitudinal cracks are visible on the pure W cathode; these small fractures are produced by the Hydrogen Embrittlement Cracking effect ( HEC ) on the tungsten.
Visit the Naudin Lab website for complete details...