The Technology of Low Temperature Carbonization
by
Frank M. Gentry
[ Chapter 5: Nitrogenous & Other
By-Products ]
Table of Contents
Preface & Table of
Contents
Chapter I ~ Fundamentals
Chapter II ~ Low Temperature
Coal Gas
Chapter III ~ Low Temperature
Coal Tar
Chapter IV ~ Low Temperature
Coke
Chapter V ~ Nitrogenous & Other
By-Products
Chapter VI ~ Processes of Low
Temperature Carbonization
Chapter VII ~ Operation, Design,
& Materials of Construction
Chapter VIII ~ Economics &
Conclusion
Bibliography
Name Index
Subject Index
[ Note: The quality of the scanned graphics (tables & figures) are "uneven" at best despite repeated efforts to scan and tweak the images. -- R.N. ]
Distribution of Nitrogen
Effect of Temperature
Ammonium Sulfate
Thermal Decomposition
Rate of Decomposition
Ammonia Oxidation
Nitrogen & Hydrogen Atmospheres
Steaming
Sulfur Distribution
Thermal Transformations
Temperature Effect
Desulfurization
According to Fieldner, Selvig, and Paul (210), the nitrogen
content of coals of the USA varies from 0.32% in a sample from
California, to 2.15% in a sample from Maryland. The Utah coal
which was coked at low temperature by Parr and Layng (101)
contained 1.4% nitrogen. Porter and Ovitz (96) found the
following percentages of nitrogen in the coals distilled by
them: Pennsylvania, 1.23%; Illinois, 1.31%; Virginia, 1.07%;
Wyoming, 1.06%; and Utah, 1.16%; while the bituminous coals
carbonized by Taylor and Porter (98) contained nitrogen to the
following extent: Pennsylvania, 1.53%, Illinois, 1.55%; West
Virginia, 1.45%; and Wyoming, 1.49%. In Table 16, the average
percentage of nitrogen found in coals of the United States has
already been given. As a rule, these coals have from 1.1% to
1.6% nitrogen, which corresponds to 24.5 pounds and 36 pounds of
nitrogen per net ton of coal, respectively. On the basis of 100%
theoretical yield, this amounts to from 30 to 43.5 lb of gaseous
ammonia per net ton, or 115 to 169 lb of ammonium sulfate per
net ton of raw fuel.
The efficiency of the nitrogen recovery depends largely upon the
process of carbonization used. When the coke is completely
gasified and the by-products recovered, as in some producer
processes, the yield of ammonium sulfate attains as much as 85
lb/net ton, which is 61.5% of the theoretical. It is interesting
to note from Table 86, which gives the average recovery of
ammonium sulfate in the three recovery methods of coal
carbonization, that the greatest efficiency of recovery is
obtained in gasworks practice and the lowest efficiency in low
temperature carbonization, metallurgical coke ovens occupying an
intermediate position. We shall see later that the reason for
this resides in the fact that the low temperature retorts
operate at too low a temperature to drive the nitrogen from the
coke in quantities and that the metallurgical coke ovens operate
at such a high temperature in order to get a good coke
structure, that the ammonia evolved is partly decomposed by the
heat.
The nitrogen is present within the coal in a number of forms.
Under destructive distillation some of it appears in the gas as
free nitrogen, some as ammonia, and some as cyanogens. A small
amount of the nitrogen appears in the tar, as nitrogen bases,
and a large proportion ordinarily remains in the coke. The
nitrogenous products found in low temperature tar have already
been fully discussed in Chapter III under the subject of tar
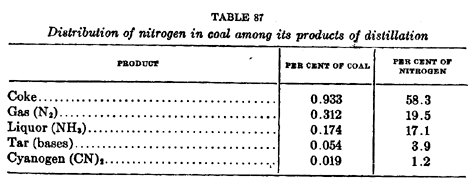
bases. Table 87, after Lewes (17), quoting McLeod, gives the
distribution of nitrogen among the volatile products and
residuum of carbonization. This table presents the interesting
fact tht the major portion of nitrogen remains in the
carbonization residuum and a large part is lost in the gases
which are evolved, so that, in nay event, hardly more than 30%
of the total nitrogen is recoverable in a useful form. It has
been shown in Table 73 that the percentage of nitrogen in the
coke after distillation is greatest for low temperature coke and
least for that made in metallurgical coke ovens.
The gas evolved in low temperature carbonization contains
considerable elementary nitrogen. Thus, Parr and Layng (101)
reported 3.87% of the gases which they obtained from Utah coals
at 750° C to consist of this gas. Davis and Parry (97) found
that the Pennsylvania coals which they examined averaged 5.83%
nitrogen in the gas. The gas evolved in the manufacture of
Coalite runs considerably higher in nitrogen content, 9.28%
having been recorded by Parr and Olin (31)

The percentage distribution of the nitrogen in coal among its products of distillation varies with eh temperature of carbonization. Simmersbach (211) studied the effect of temperature upon the distribution of the elementary nitrogen with the results shown in Figure 41. These experiments were made upon Silesian coal with a total nitrogen content of 1.39%. Reference to the illustration shows that the maximum percentage of nitrogen occurs in the ammonia at 900° C, while the minimum percentage of nitrogen in the gas occurs a hundred degrees lower. Heating of the coal beyond 900° C decreases the nitrogen remaining in the carbonization residuum but does not increase the percentage of ammonia. The additional nitrogen evolved merely goes to increase its content in the gas. The nitrogen content of the tar remains about constant, irrespective of the temperature beyond about 600° C. It is interesting to note that, even at a temperature of 1200° C, over a quarter of the nitrogen contained in the coal remains in the coke.

We have already seen in Table 25 the variation in the proportion of free nitrogen in the gas which was evolved from Lancashire coal distilled up to 400° C by Burgess and Wheeler (104). Lignites and peats usually contain usually contain much more nitrogen than bituminous coals and a large part of it appears as free nitrogen in the gas upon destructive distillation. We have seen in Table 27 that as much as 20% of the gas which was evolved from the carbonization of peat in vertical retorts was free nitrogen. Again, in Table 29 the results of Benson and Canfield (108), on the distillation at various temperatures of Newcastle lignite from the state of Washington, show as much as 42.7% free nitrogen in the low temperature lignite gas. After 400° C temperature of carbonization in this case, there is apparently no increase in the percentage of free nitrogen in the gas, at least up to 600° C.
In Figure 13, it is seen from the results of Taylor and Porter (98) in heating Wyoming coal for long periods at 350° C, that after 25 hours of distillation, practically all ammonia had been removed from the coal, whereas the production of saturated hydrocarbons, hydrogen, and the oxides of carbons continued even to coking periods of 200 hours. Consequently, the longer the period of distillation at this temperature, the less the proportion of ammonia to be expected in the gas.
Giles and Vilbrandt (99), in their distillation of Farmville (NC) coal at temperatures ranging from 200° C to 660° C, found that the ammonium sulfate produced, expressed as percentages of the original coal, amounted to 0.17% at 200° C, 1.23% at 300° C, 2.2% at 420° C; 2.25% at 540° C; and 2% at 660° C. Analyses of the semi-coke obtained by Benson and Canfield (108), when distilling a Washington lignite at temperatures ranging from 150° C to 600° C, showed the carbonization residuum to contain 2.9% nitrogen at 450° C, and 0.5% at 600° C.
The authorities differ so greatly in the yields of ammonium sulfate which they have obtained in their experiments on low temperature carbonization that a tabulation of their average results, given in Table 88, wills serve valuable comparative purposes. The average of the yields given in this table is almost exactly 14 pounds of ammonium sulfate per gross ton of coal, which is the value recommended by the Fuel Research Board, after their extensive experiments on many different coals, as the most reliable yield to be expected in low temperature carbonization. In order to pay for recovery, the ammoniated liquor must be approximately 1.7% ammonia or 8 ounce strength. This means a recovery of 14 pounds per ton. Otherwise, it is necessary to scrub the primary liquor with fresh gas until it reaches that concentration, which of course means a larger operating expense and its consequent reduction of profit.
It is quite obvious from Table 88 that, with one exception and within the temperature limits of low temperature carbonization, the higher the temperature the greater is the yield of ammonium sulfate. The experiments of Burgess and Wheeler (104) on the carbonization of Silkstone coal at temperatures ranging from 450° C to 900° C, as given in Table 23, show that at the temperatures of 500° C to 700° C inclusive, about 1.5% of the gas evolved consists of ammonia. At 750° C, which under these circumstance appeared to be extremely favorable to the formation of ammonia, there was a copious evolution, which thereafter decreased with temperature rise.
Table 89 gives the temperature variation of ammonium sulfate yield, as determined from the experiments of Lewes (17) and of Simmersbach (211). The later used Silesian coal, which is by no means typical, for the yields quoted by Simmersbach are much higher than ordinarily obtained between 600° C and 900° C. The figures given by Lewes, who used a good gas coal, are perhaps more representative of ordinary coals. The yields are from charges subjected to carbonization over a period of 6 hours. These figures are more than 30% lower than those given by Simmersbach in the low temperature range, but they are more representative of the conditions obtaining in full-scale operations.



Figure 42 gives the data of Table 89 in pounds of ammonium sulfate per gross ton of coal. In the illustration, it is seen that the yield of ammonium sulfate reaches a maximum at 900° C, according to both authorities. An average bituminous coal may be said to yield considerably less ammonium sulfate when carbonized at low temperatures, from 500° C to 700° C, than when distilled at high temperatures over the range of 900° C to 1100° C. It will be noted in the illustration that the curves showing the yields of ammonium sulfate decrease after attaining their maximum. It will be pointed out shortly that this drooping of the yield curve is the direct result of decomposition of the ammonia. Temperature serves to drive the remaining nitrogen from the coke, as indicated by Table 89 and Figure 42, but not so much in the form of ammonia as in the form of elementary nitrogen, that is, in the absence of protective gaseous atmospheres.
That the yield of ammonia attains a maximum at a certain temperature under given conditions and thereafter decreases as the temperature of carbonization rises, has already been demonstrated. The reduction in the percentage of ammonia in the gas when it is subjected to superheating has also been noted in Table 36, as well as the decomposition of ammonia effected by passing the gas over a small incandescent surface, as shown in Table 37, and by the catalytic action of hot brick contact surfaces, recorded in Table 38. Presently, the subject of ammonia decomposition by active brick surfaces will be taken up again.
A study of the equilibrium conditions surrounding the chemical equation,
[22] 2 NH3 => N2 + 3 H2
will explain this phenomenon clearly. This reaction, when proceeding from right to left, is an exothermic one and takes place with the liberation of 24,000 calories of heat (116). We can predict immediately, therefore, from thermochemistry, that low temperatures are favorable to the stability of ammonia.
The effect of temperature variation on ammonia decomposition was investigated by Ramsay and Young (214). Their first experiments consisted in passing the gas through a porcelain tube, filled with broken pieces of the same material. In the second series of experiments, an iron tube was substituted for the porcelain tube for the purpose of investigating the effect of the tube material as a catalytic agent. The results are plotted in Figure 43. It is seen from the illustration that decomposition of the ammonia begins below 500° C, attains a maximum rate from 600° C to 700° C, and is practically complete at 800° C. The catalytic effect of the surface of the tube is very pronounced. Thus, iron increased the decomposition at a given temperature often as much as 100% over that obtained in the presence of porcelain. It must be remembered that the equilibrium of Equation [22] is a dynamic one and the effect of a catalytic agent is to accelerate the rate of reaction in one of the directions, the net result being an increase or a decrease in the percentage of ammonia which may exist under given thermal conditions.
The data in Figure 43 apparently disagree with the results of Simmersbach and of Lewes given in Table 89 and in Figure 42. The maximum yield of ammonia occurs at 900° C in Figure 42, which is beyond the temperature prescribed in Figure 43 for complete dissociation of the ammonia. Presently, we shall see that this can be readily accounted for by the retarding influence exerted by certain other constituents of the gas. In any case, the quantity of ammonia yielded is considerably less than the actual amount formed and the reduction of the yield at high temperatures must be attributed to its decomposition. The nitrogenous matter in coal is doubtlessly present in many forms. Some of the constituents of the coal give ammonia on simple distillation, while others react with the moisture and hydrogen which is present to form ammonia-yielding compounds.

Foxwell (215, 216) made an extended study of the thermal dissociation of ammonia in coke ovens. He used in his experiments a silica reaction tube coated with a thin layer of carbon. Using such a tube packed with coke, Foxwell studied the concentration of ammonia at various temperatures and found that the decomposition was a second order reaction whose rate of reaction, therefore, was expressible by the equation,
[23] dx /dt = k (a-x)2
where x is the amount of ammonia decomposed in time, t, and a is the initial concentration of ammonia expressed in millimeters of mercury partial pressure. From Equation [23] we may solve for the velocity constant, k, as follows:
[24] k = 1 / t ( 1 / a-x - 1 / a)
Despite the heterogenous reaction, occurring in the thermal dissociation of ammonia in coal gas, it was found that a bimolecular coefficient adequately expressed the results, even though it is difficult to interpret its meaning. Perhaps the presence of coal gas and water vapor exerted an influence which disguised the true character of the reaction.
The temperature variation of the reaction velocity constant was found to agree with Arrhenius' empirical equation,
[25] k2 = k1 E A ( 1 / T2 - 1 / T2 )
where A = 13,300 and K2 and K1 are the constants at temperatures T2 and T1 respectively. The calculated and measured values of the velocity constant, k, for various temperatures, when coke is used as the contact material, are given in Table 90, from which it is seen that the temperature increase of the velocity of the reaction is comparatively slow. Foxwell (215, 216) found that the ammonia decomposed to some extent below 600° C, contrary to the findings of other investigators.
It has been noted in Table 38 that hot brick surfaces catalyze the decomposition of ammonia an it has been pointed out that, since the equilibrium given in Equation [22] is dynamic, the effect of a catalyst is merely to accelerate the reaction rate which must be reflected in a variation of the coefficient, k, of Equation [23] and Equation [24]. This is indeed the case. Table 91 gives the analyses of four different bricks, two of which are silica bricks and two of which are siliceous bricks. The table gives for each brick a sample low in ferric oxide and a sample high in ferric oxide. The great influence of silica and ferric oxide on the velocity constant, k, is quite apparent. It can be concluded that silica brick has a much smaller decomposing influence than siliceous brick and that, from the standpoint of preservation of ammonia, the presence of iron is highly undesirable. All the brick samples had substantially the same porosity. Later we will discuss the results of Mott and Hodsman (217), who studied the influence of contact materials on the decomposition and oxidation of ammonia in various gaseous atmospheres.
Since brick contains many of the oxides found in coal ash, it might be anticipated that the constitution of the ash would have an important bearing on the decomposition of ammonia evolved from the coal. Under the subject of catalysis in Chapter I, it has already been pointed out that Lessing (81), Lessing and Banks (82), and Marson and Cobb (83) investigated the effect of ash analysis on the solid residuum from distillation. Foxwell (215, 216) made a number of experiments to define the influence of coke ash on the ammonia decomposition velocity constant. He used a Durham coal, containing 8.1% ash in his experiments, to which various ingredients were added in different samples for the purpose of ascertaining the effect of each substance. The analyses of the ash from 8 specimens is given in Table 92, along with the mean velocity constant for each sample at 755° C. Sample No. 1 was the ash of a gas coke alone; Sample No. 2 was the as of a Durham coal alone; Sample No. 3 contained an addition of 5% impure pyrites from a coal seam; Sample No. 4 contained an addition of 3% rutile; Sample No. 5 contained an addition of 5% orthoclase felspar; Sample No. 6 contained an addition of 5% ferric oxide; Sample No. 7 contained an addition of 3% ignited lime; and Sample No. 8 contained an addition of 10% ignited lime.
The velocity constant given in Table 92 is the mean value for periods of contact ranging form 0.78 second to 4.24 seconds, within which time intervals the constant was found to be independent of the time of contact. Examination of the table discloses that rutile and felspar have practically no catalytic effect, whereas iron in certain forms and lime have a most vigorous influence. During carbonization, pyrites and ferric oxide are converted to ferrous sulfide and metallic iron, respectively. Ferrous sulfide increases the reaction rate only slightly, whereas the addition of about 3.75% metallic iron, reduced form ferric oxide, increased the reaction rate 15-fold. Consequently, if iron is present in the coal as pyrite, its catalytic effect will be negligible, but if it is present as ferric oxide, the decomposition of ammonia will be tremendously accelerated. The result of the addition of lime was not anticipated and is somewhat inexplicable. The addition of lime is a well known practical method of increasing ammonia yield in carbonization practice, but these experiments showed that the addition of 3% lime more than doubled the rate of dissolution, which was further increased only slightly by larger additions of the same material.
Some significant results were obtained by Foxwell (215, 216) when 1% salt was added to the coal. In this case, the value of the reaction velocity constant, k, did not remain constant when the period of contact varied, as in the case of materials previously tested. As the period of contact increased, the value of k decreased in the following manner: for 1.14 seconds, k = 0.00200; for 1.20 seconds, k = 0.00142; for 1.33 seconds, k = 0.00131; and for 3.86 seconds, k = 0.00093. These data show that when the period of contact is long enough, the salt acts as a negative catalyst and retards the dissociation of ammonia. The influence of salt is attributed to its reaction with water vapor and carbon dioxide to form sodium carbonate with the liberation of hydrochloric acid, the latter being the retarding agent. Explanation of the decrease in velocity constant, as the period of contact is lengthened, has been suggested to reside in the disappearance of the hydrogen chloride, as time progresses, by its reaction with the ash constituents to form hydrogen sulfide, ferrous chloride, and other compounds. The presence of hydrogen sulfide in the gas was indicative of this explanation. Foxwell (215, 216) attempted to ascertain the effect of hydrochloric acid concentration on the ammonia decomposition velocity constant and obtained some results that are difficult to interpret. He found that the velocity constant decreased from about 0.0030 to about 0.00060 as the ration of hydrogen chloride to ammonia concentration increased from zero to 0.1, at which point there was an abrupt change. Thereafter, the velocity constant rose to a maximum of about 0.0015 at a concentration ratio of 0.4, finally decreasing to about 0.00035 at a concentration ratio of unity. The decrease up to a concentration ratio of 0.1 is explained by the negative catalytic effect of hydrogen chloride; the rise to a maximum at a concentration ratio of 0.4 is attributed to the positive catalytic effect of metallic iron, produced by the reaction of hydrochloric acid with ferrous sulfide to form ferrous chloride, which in turn reacts with moisture to form ferrous oxide, the latter being finally reduced by hydrogen to metallic iron; while the reduction of velocity constant up to a concentration ratio of unity can be conceived as the result of the formation of ammonium chloride, which is stable at temperatures above 800 C, thus decreasing the effective quantity of ammonia available for dissociation.
An extensive investigation on the factors which influence the destruction of ammonia by oxidation in the carbonization of coal was carried out by Greenwood and Hodsman (218, 219). A knowledge of this phenomenon is clearly important, because of the likelihood of indrawn air in a leaky retort and because of the part played by water vapor and materials of construction in retarding or promoting ammonia oxidation. It is very well known that the oxidation of ammonia is exceedingly sensitive to catalysts, both with respect to the velocity of oxidation and with respect to the products of the reaction. Ammonia may react with oxygen to yield elementary nitrogen and water, or it may react to form nitric oxide and water. Under the conditions obtaining during carbonization, the formation of gaseous nitrogen upon oxidation is the more likely of the two to occur, but conceivably, by the use of certain catalytic agents, the ammonia could be oxidized almost quantitatively to the oxide. Apparently, ammonia is completely unstable in the presence of oxygen at all temperatures, so that neither of the above mentioned reactions is reversible under ordinary, or indeed under any known, conditions. Consequently, the decomposition of ammonia by oxidation cannot be regarded as limited by conditions of equilibrium or by the law of mass action. Under given physico-chemical conditions with a given catalyst, there is always a temperature at which the tendency to form nitric acid from the reaction of ammonia and oxygen is at a maximum. Above and below this optimum temperature the tendency is to form free nitrogen, probably as a result of the interaction of nitric oxide that is produced and fresh ammonia to form elementary nitrogen and water. This sufficiently accounts for the fact that nitrates and nitrites are never found in the gas liquors.
Greenwood and Hodsman (218, 219) observed that the catalytic effect of a porous firebrick which contained 1.4% of ferric oxide decreased from a marked activity, in the initial passage of gas, to about 25% of its former value after prolonged flow of gas. Thus, its effectiveness diminished from about 82% of the ammonia oxidized, after 10 liters of gas had passed over the catalyst, to about 25% decomposed after 120 liters had flowed through the reaction tube, the decay of activity apparently following an exponential law. This fatigue of the brick catalyst cannot be attributed to poisoning by other constituents of the gas, for the same phenomenon was observed when pure air was passed. The author suggests than an explanation may be found in surface adsorption and occlusion of gas within the minute pores.

Greenwood and Hodsman (218, 219) passed ammonia, mixed variously with dry and moist air, over different contact materials for quite a range of temperatures with the results illustrated in Figure 44. A blank experiment was made with the ammonia mixed with dry air and passed over silica, representative of a relatively inactive contact material, to determine the temperature range over which oxidation takes place when catalysis is minimized. Below 500° C, very small quantities of nitrous acid, but never nitric acid, were detected, while above that temperature only free nitrogen was produced by the reaction. The introduction of a nitrogenous atmosphere, containing from 1% to 2% oxygen, caused a retardation of oxidation below 700° C, due to the reduction in oxygen concentration, but above that temperature this effect was not noticeable. When an atmosphere of coal gas containing 1% to 2% oxygen was substituted in place of air, with silica as the contact material, oxidation of ammonia was so effectively retarded that it was zero at 600° C and less than 15% had been destroyed up to 800° C. When firebrick was substituted for silica as the contact material, in the presence of coal gas there was only a slight increase in oxidation. On the other hand, when fireclay was used as the catalytic agent in air, the oxidation of ammonia was tremendously accelerated, as seen in Figure 44. From zero decomposition at 450° C with silica, the oxidation was raised to about 8% with fireclay and from about 25% oxidation at 750° C with silica to about 70% with fireclay. When, however, moist air was substituted for dry air in the presence of fireclay contact material, there was a decrease of about 20% in the proportion of ammonia oxidized. All of the experimental data were reduced to a contact time of 7 seconds.
The results indicated that, under the conditions of the experiment up to temperatures of 800° C, there was very little oxidation of ammonia in the presence of coal gas, but that at the end of the passage the entire oxygen present had been removed, presumably by combination with the hydrogen or by reaction with the hydrocarbons of the coal gas. Had the entire ammonia present been oxidized, only a fraction of the oxygen would have been absorbed in this reaction. Even at 600° C, absolutely no ammonia was oxidized in an atmosphere of coal gas containing oxygen, although at this temperature the oxidation amounted to about 12% and about 8% in atmospheres of air and nitrogen, respectively. From these experiments, it is apparent that when ammonia is heated with oxygen in the presence of coal gas, oxidation of the hydrogen and hydrocarbons takes precedence over oxidation of the ammonia, and, if any of the latter is destroyed at all, it must be attributed to thermal dissociation rather than to oxidation.
The presence of moisture when a mixture of air and ammonia was passed over silica had very little effect, decreasing the oxidation but 2.5% at 750° C. The presence of moisture in coal gas caused no difference whatever in the amount of ammonia oxidized. However, it is seen from Figure 44 that when moisture was admitted to the mixture of air and ammonia there was a material retardation in oxidation with fireclay as the contact material. The addition of 2.8% water vapor caused a reduction of the percentage ammonia oxidized at 740° C from 64% to 52.3%. The influence of water vapor on the stability of ammonia in the presence of oxygen can be attributed to two factors, viz: the increased rate of gas flow with the consequent shortening of the period of contact with the active surfaces and the specific effect of the water vapor. The curves in Figure 45 show severally the variation of oxidation at 740° C with the rate of flow for a mixture of ammonia and dry air; the variation of oxidation at the same temperature, as a function of the rate of flow, when the air is saturated with moisture; and, by subtraction, the resultant curve, which may be regarded as showing the specific action of the water vapor. The preservative influence of moisture on the oxidation of ammonia was marked only in the case of contact surfaces exhibiting active catalytic effects, and then the activity of the catalyst was only partially retarded. The most plausible explanation of this behavior is found in the supposition that a unimolecular film of moisture forms on the catalytic surface, thus restricting the access of ammonia and oxygen to the active material.
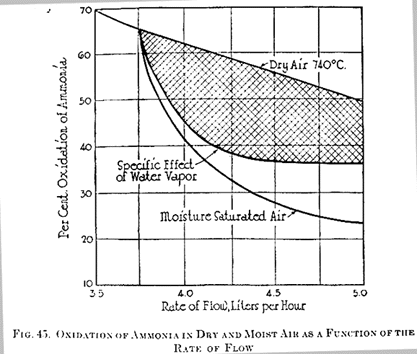
Mott and Hudson (217) investigated the influence of water vapor, and also the character of the contact materials, on the decomposition of ammonia at various temperatures. Their experiments were performed with a mixture containing about 1.5% ammonia, which is the same order of concentration for that gas found in carbonization practice. Figure 44 shows the decomposition of ammonia by oxidation as a function of the temperature when heated with dry and moist air and passed over chattered brick which had previously seen hard service in ovens where salty coals were coked. This brick had been badly corroded, iron from the coal ash having been volatilized as the chloride and deposited within the brick for some distance as the metal or as the oxide. Analysis showed this brick to contain 79.84% silica, 11.49% alumina, 1.31% titania, 3.95% ferric oxide, 0.6% lime, 0.12% magnesia, and 2.47% alkalies, by difference. The high iron content of this brick probably accounted for its high catalytic action. These curves should be compared with those in Figure 44 for a Farnsley brick, designated as fireclay, from which it is observed that the iron in the chattered firebrick doubtlessly deposited as the more active metallic form, whereas in the fireclay it probably existed in combination with silicates. The presence of about 3% water vapor lower the oxidation at a given temperature much more than could be accounted for by reduction in time of contact, thereby confirming previous experiments indicating a specific action of water vapor.
Nitrogen & Hydrogen Atmospheres ~
Mott and Hodsman (217) continued their work on the effect of contact surfaces on the oxidation and dissociation of ammonia in different atmospheres. The results in atmospheres of coal gas, hydrogen, and nitrogen, with and without the presence of oxygen, are shown in Figure 46. In these experiments the chattered firebrick previously referred of was used as the contact material. The data were all calculated to the same period of contact.
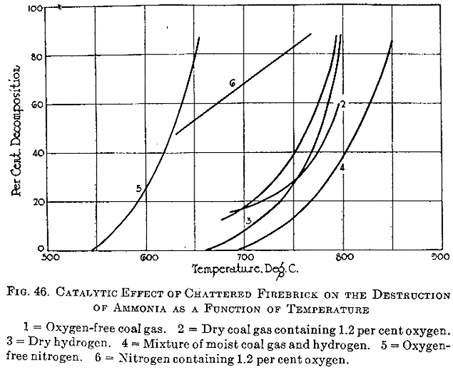
When a mixture of dry hydrogen and ammonia, to the extent of about 1.5%, was passed through the reaction tube, dissociation began at about 660° C and rose rapidly until at 800° C it was practically complete. Under these conditions the velocity of dissociation increases rapidly with the temperature, the velocity constant k of Equation [23] increasing from 0.00074 at 675° C to 0.0059 at 750° C and to 0.081 at 800° C. At 700° C the decomposition amounted to about 9.5%; at 750° C to about 26%; and at 800° C to about 86%. When a nitrogenous atmosphere was substituted in place of hydrogen, dissociation began as low as 550° C and reached nearly 80% at 650° C. When 1.2% oxygen mixed with nitrogen was used, the dissociation curve was apparently a linear function of the temperature, increasing regularly from about 46% at 625° C to about 90% at 775° C. The dissociation of ammonia is tremendously greater in the presence of nitrogen than in atmospheres of hydrogen, the two curves being roughly parallel and about 150° C apart. In nitrogen the ammonia was almost completely decomposed at 660° C, whereas decomposition was just measurable at that temperature with hydrogen. It could of course be argued, from the law of mass action, that changes in the hydrogen concentration would be far more effective in preserving the ammonia than changes in the nitrogen concentration. This, of course, is true near the equilibrium condition, but it is neither necessary nor probable under conditions where the reverse reaction is negligible. Quite surprisingly, the presence of 1.2% oxygen in the nitrogen above 650° C has a preservative influence on the ammonia. Whether or note this beneficial result is due to initial dissociation and reaction of the hydrogen to form water vapor with its inhibitory influence, or to an entirely different mechanism, such as direct oxidation, is a matter of conjecture. Finally, when an atmosphere of dry coal gas was used, the destruction of ammonia amounted to 18% at 700° C, and was about complete at 800° C; but when 1.2% oxygen was present in the gas, the decomposition was only 62% at the higher temperature. Here the benefit derived from the presence of oxygen is clearer, for water vapor is doubtlessly formed by the preferential oxidation of hydrogen in the coal gas.
Further experiments by Mott and Hodsman (217) on the decomposition of ammonia, when passed over coke, gave results similar to those derived from the use of chattered firebrick as the contact surface, but the decomposition at a given temperature under a given atmosphere was considerably less, showing the coke to be a far more inactive catalyst than the chattered brick. They concluded that since carbon has been shown to be an indifferent catalytic agent, the quantity and quality of the coal ash remaining in the coke is more likely to influence the decomposition of the ammonia. We have already seen that Foxwell (215, 216) demonstrated that the decomposition of the ash, indeed, had a great effect on ammonia dissociation.
Monkhouse and Cobb (220) observed that, of the total nitrogen present in the coal, only 10% to 25% is obtained as ammonia, the greater portion, amounting to 40% or 80%, remaining in the coal and the rest being found in the gas as free nitrogen or as cyanogens. Many factors, such as temperature of distillation, nature of the coal, and rate of carbonization, affect the liberation of ammonia during coking. Temperatures above 500° C are necessary for a good yield of ammonia and the higher the temperature, the less nitrogen remains in the coke. In general, the older the geologic age of the fuel, the less the proportion of total nitrogen obtained as ammonia. Finally, the slower the rate of distillation, the more nitrogen is evolved as ammonia, provided the vapors are quickly removed.


In their study of the liberation of nitrogen from coke as ammonia, Monkhouse and Cobb (220) used a Yorkshire bituminous coal which contained 5.4% moisture, 34% volatile matter, 56% fixed carbon, and 4.7% ash. The ash consisted of 25.1% silica, 33.5% alumina, 10.1% ferric oxide, 13.1% lime, 12.9% sulfuric oxide, and 5.1% alkali oxides. Ultimate analyses of the raw coal and the three cokes obtained from it are shown in Table 93. The 500° C coke resembled gas coke, and the 1100° C coke was comparable to byproduct oven metallurgical coke. The table shows clearly that the nitrogen present in the carbonization residuum was reduced as the temperature advanced. The ammonium sulfate equivalent of nitrogen in the raw coal was 176.3 pounds per gross tone of coal; that in the 500° C coke was 197.5 pounds per gross ton of coke, or 133.3 pounds of ammonium sulfate per gross ton of coal; the 800° C coke contained nitrogen equivalent to 11.5 pounds of ammonium sulfate per gross ton of coke, or 66.5 pounds per gross ton of coal; while the 1100° C coke had 61.2 pounds of equivalent ammonium sulfate per gross ton of coke, or about 34 pounds per gross ton of coal. These cokes were heated at various temperatures in different atmospheres to determine the specific action of different action of different gases on the production of ammonia.
Table 94 shows the results of heating the different representative cokes in the presence of nitrogen. The amount of ammonia evolved from the 100° C coke under these conditions was so small as to be negligible. The 500° C coke under these conditions had 1.34% nitrogen remaining after reheating to 1000° C, while the 800° C coke had 1.02% remaining after reheating to the same temperature. This table should be compared with Table 95, which gives a nitrogen balance for the tests of these cokes when they were heated to the final temperature of 1000° C in an atmosphere of nitrogen. Reheating the 800° C coke in stages to 1000° C removed 30% of the coke nitrogen, but if any ammonia was formed, practically all of it was decomposed under these conditions, as only 0.2% was recovered.
Monkhouse and Cobb (220), with the idea of minimizing dissociation, next determined if any more ammonia could be obtained from the low temperature coke by heating it in stages to 800° C, as compared to heating it directly to 800° C in an atmosphere of nitrogen. The results indicated that the ratio of ammonia to free nitrogen was slightly greater in the multi-stage heating than in direct heating, so that there was some little reduction of dissociation.




Monkhouse and Cobb (220) also reheated the cokes obtained by them at different temperatures in an atmosphere of hydrogen, thereby obtaining a distinctly different result from the use of nitrogen. Table 96 gives the results of heating the 500° C and 800° C cokes in stages to 1000° C in the presence of hydrogen and Table 97 gives a nitrogen balance for the same conditions. The 1100 C coke was heated in hydrogen up to 1000° C, but no ammonia whatever was evolved. The nitrogen remaining in the 500° C coke, after treatment up to 1000° C, was 0,65% and that remaining in the 800° C coke was 0.84%.
The illustration in Figure 47 shows graphically the difference between the effect of nitrogen and hydrogen atmospheres on the production of ammonia by reheating the low temperature coke to drive off the nitrogen. At 1000° C in an atmosphere of nitrogen, the ammonia recovery ceased to increase appreciably with rising temperature, due undoubtedly to the increased rate of dissociation. The marked influence of a hydrogenous atmosphere on the yield of ammonia is seen by the fact that, when the low temperature coke was reheated at 1000° C in an atmosphere of hydrogen, 34.2% of the nitrogen in the coke was recovered as ammonia, with 28.2% of the nitrogen originally present remaining in the coke residuum, as compared with 11.7% recovered as ammonia in an atmosphere of nitrogen, with 60.4% of the original nitrogen remaining in the coke after treatment. In all cases, the rate of ammonia recovery was more rapid when the various cokes were reheated at 800° C than at either the lower temperature of 600° C or at the higher temperature of 1000° C. If hydrogen aided the formation of ammonia in nitrogenous compounds evolved at the higher temperatures, it was rapidly decomposed. It is quite conclusive that hydrogen exerted on the coke a specific action which favored the formation of ammonia, as compared with the indifferent action of such inert atmospheres as nitrogen.
One remaining phenomenon has its influence on the quantity of ammonia yielded. That is what has been called the secondary production of ammonia by the action of hydrogen gas on the incandescent coke. Tervet (222), in his experiments, passed a current of hydrogen over incandescent coke and found that the ammonia yield was increased 100%. The action of hydrogen on the soft coke is such as to attack the nitrogen which it contains and to form increased quantities of ammonia. Hydrogen did not attack hard coke at all and the effect on medium coke was much less pronounced than when the soft variety was used. A yield as high as 94 pounds of ammonium sulfate per gross ton was reported upon carbonizing the charge of soft coke slowly at 1000° C in an atmosphere of hydrogen. It seems probable, therefore, that over the range of 500° C to 800° C, hydrogen gas has a specific action capable of liberating additional quantities of ammonia, as well as of acting as a preservative of the ammonia otherwise formed.
The use of steam during carbonization is well known to increase
greatly the yield of ammonia in high temperature processes, but
its effect in low temperature methods is questioned among the
authorities. The Fuel Research Board maintains that here again
the principal function of the steam is to sweep the retort clean
of gaseous products, thus removing the ammonia as quickly as
possible from the conditions favorable to its decomposition.
They maintain that little or no increase in nitrogen, evolved as
ammonia, can be expected through the use of steam in low
temperature processes, although in coals of low moisture content
it might assist in the recovery of small quantities of ammonia
which would remain uncondensed and make scrubbing necessary. We
have already seen in Table 43 the influence of increasing
percentages of steam on the analyses of low temperature gas, the
nitrogen content being decreased from 13.8% in dry distillation
to 7.6% when 20% steam was passed into the retort. This
reduction in free nitrogen in the gas doubtlessly arises from a
reduction of the thermal dissociation of ammonia.
Porter and Ovitz (96), in comparing the yield of ammonium
sulfate obtained from dry and moist coals, substantiated the
opinion of the Fuel Research board that high moisture content
increases the efficiency of ammonia recovery. They found that,
when computed upon the basis of dry coal, a yield of 25.3 pounds
of ammonium sulfate was obtained per gross ton of dry coal, as
compared with 26.9 pounds of ammonium sulfate recovered when the
same coal was carbonized while moist.
The experiments of Mott and Hodsman (217) demonstrated that the presence of 25% steam at 850° C completely prevents dissociation of the ammonia, which ordinarily is completely decomposed at a temperature 50° C lower. At 785° C they found that 72% of the ammonia ordinarily dissociated, but with 12.5% water vapor only about 8% decomposed.
Davis and Parry (97), using Pennsylvania coal of the Freeport bed, found that the use of 88% steam during carbonization at 550° C increased the yield of ammonium sulfate 43%. The variation of ammonium sulfate yield at various temperatures under steam distillation, as determined by them, is given in Table 98. At 550° C, without steam, they obtained only 10 pounds of ammonium sulfate per net ton, the gas evolving containing 8.2% nitrogen and 1.9% nitrogen remaining in the coke.

It is seen in Figure 46, for the experiments of Mott and Hodsman (217), that when 1% to 2% ammonia was passed through the reaction chamber in an atmosphere of coal gas containing 1.2% oxygen, the oxidation amounted to about 17% at 700° C, and to about 41% at 750° C, whereas when oxygen-free coal gas was used the decomposition was 18% and 61% at the corresponding temperature. At first sight, this appears astonishing, but reflection shows that in every case where oxygen was present in the coal gas it had entirely reacted with the hydrogen and hydrocarbons to form water, which undoubtedly exerted an inhibitory effect on the ammonia decomposition, it having already been demonstrated that under such conditions the oxidation of ammonia is prevented by the preferential oxidation of the hydrogen and the hydrocarbons. When a mixture of coal gas and hydrogen, containing about 3% moisture, was used, the inhibitory effect of water vapor was even more marked than in the case of an atmosphere of air, as shown by the two upper curves in Figure 44. At 700° C, the presence of 3% water vapor reduced the ammonia decomposition from about 17% to about 3% and at 800° C from about 95% to about 38%. The reduction in the time of contact, through presence of steam, could account for a difference of only about 2%.
Monkhouse and Cobb (220), having ascertained that reheating the 1100° C coke in dry hydrogen up to 1000° C was without effect in removing the nitrogenous material of the coke in the form of ammonia, investigated the influence of using a mixture of steam and hydrogen, the former being present to the extent of 65%. The results are shown in Figure 48, from which it is seen that the presence of moisture caused a slow but steady evolution of ammonia up to 800° C. At 1000° C the evolution of ammonia increased greatly, doubtlessly because of gasification, according to well established principles of ammonia production with excess steam, as used in the Mond system. It has been stated that in such reaction the carbon-nitrogen ratio of the fuel remains constant during gasification, which demonstrates that the production of ammonia in gasification processes with the sue of steam is accompanied by simultaneous consumption of the carbon surrounding the nitrogenous material.


The successive action of nitrogen, hydrogen, and steam was tried on low temperature coke to ascertain how far the nitrogen remaining in the coke from one treatment could be removed by application of gaseous atmospheres of more specific action. The experiments were made on 500° C low temperature coke at a reheat temperature of 800° C. heating was continued with each gaseous atmosphere until the evolution of ammonia practically ceased. Two curves, giving the results of these experiments, are reproduced in Figure 49, one for successive heatings with nitrogen, hydrogen and steam, and one for only nitrogen and steam. At the end of the nitrogen stage, 10.1% of the coke nitrogen had been yielded as ammonia; 19.6% was evolved as free nitrogen; and 70.3% remained in the coke residuum. When hydrogen was then introduced, an additional 32% of the nitrogen was gasified, of which 31% appeared as ammonia, the decomposition being practically nil under these conditions. Steam diluted with 37.5% nitrogen was next admitted to the retort. Due to the water gas reaction, the quantity of gas yielded in this case exceeded that of the gas admitted; in fact, when the experiment was discontinued, it was found that this reaction had progressed t complete gasification, so that only the coke ash remained. In this final stage 38.3% of the nitrogen originally present in the coke was removed, 38.1% being ammonia and the rest free nitrogen. It is clearly seen that large additional quantities of the coke nitrogen can be removed by heating in atmospheres of hydrogen and of steam after treatment in nitrogen and, furthermore, that removal of the ammonia in hydrogenous and steam atmospheres is accompanied by very little decomposition. The latter is attributed to the fact that the ammonia present was so diluted by the other gases that equilibrium was not attained. Very little ammonia would have survived at 800° C if equilibrium conditions at that temperature had been reached. As a matter of fact, the concentration of ammonia itself plays a more important part than that of either hydrogen or nitrogen on the rate of ammonia decomposition at a given temperature, the rate of decomposition being higher, the higher the concentration. Figure 49 also shows the effect of omitting the hydrogen stage, the same amount of ammonia being obtained by this procedure in 40 hours of treatment as was removed in 240 hours when the hydrogen stage was introduced.
In later research, Monkhouse and Cobb (221) investigated the use of hydrogen and steam in obviating the formation of free nitrogen instead of ammonia, when the same cokes as used before were heated. Figure 50 shows the effect of reheating 500° C low temperature coke in 40-hour stages in two gaseous atmospheres. The steam contained 40% nitrogen as the carrier gas. After 40 hours of heating in the presence of steam at the final temperature, only ash remained, the carbon having been completely gasified. At the end of the 800° C stage, 9.1% of the nitrogen originally present had been evolved as the uncombined gas when hydrogen was used, as compared wit only 1.7% at the same temperature when steam was used, practically no dissociation of ammonia occurring in the latter case.


A careful examination of Figure 50 shows that, whereas the evolution of ammonia in hydrogen at a given temperature had a gradually declining rate as time went on, the evolution of ammonia in steam proceeded at a constant rate after the initial rapid evolution at the beginning of each temperature stage. In the former case, the curves are logarithmic in shape, while in the latter case they are hyperbolic. As the temperature is increased at each stage, there is an initial large evolution of ammonia which accompanies the temperature rise, even in an inert atmosphere. Simultaneously, there is taking place a gasification of the coke carbon by the water gas reaction, which is of secondary importance at the beginning of an increased temperature stage, but which ultimately becomes the controlling reaction and accounts for the characteristic linear portion of the steam atmosphere curve. In Figure 51 is shown an enlarged curve of the lower part of Figure 50, being the evolution of ammonia from low temperature coke at 600° C in various atmospheres. It is seen that, due to the characteristic shape of the steam curve, it falls below that of hydrogen for the first 35 hours of heating at this temperature. At temperatures above 700° C, the steam curve will fall entirely above the others, as the temperature becomes more favorable to the water gas reaction.
An experiment conducted by Monkhouse and Cobb (221) on the use of nitrogen saturated with 63% steam in effecting the removal of nitrogen from 1100° C high temperature coke, showed that when the reheat temperature was 800° C, only 1.36% of the coke nitrogen was removed, whereas 95.4% evolved as ammonia at 900° C. After about 90 hours treatment, 10 hours of which were at 800° C, 70 hours of which were at 900° C, and the remaining 10 hours at 1000° C, 97.3% of the coke nitrogen was removed as ammonia and 2.7% as free nitrogen. The liberation of ammonia from high temperature coke, in the presence of steam at a given temperature, was much slower than from low temperature coke, due to the great reactivity of the 500° C coke and to its ease of gasification.
Analyses of many coals of the United States by the US Bureau of Mines, as compiled by Fieldner, Selvig, and Paul (210) show that the amount of sulfur present ranges from as low as 0.11% in the case of a sample from Alaska, to as much as 11.68% for a specimen from the roof of a vein n Colorado. A great many of the bituminous coals of the USA have from 0.5% to 4.5% sulfur, the average sample containing approximately 2%. The disadvantages of high sulfur content in coke are well known. When used for domestic purposes, the fumes liberated upon combustion are objectionable to smell and are injurious to property. When coke is completely gasified, the sulfur again presents the same difficulties, for it appears as hydrogen sulfide in the gas. We have already seen in Table 32 and Table 33 that, even at temperatures as low as 450° C, considerable quantities of hydrogen sulfide may be found in the gas from low temperature distillation.
Both inorganic and organic sulfur occur in coal. The inorganic sulfur is present mostly in combination with iron, either as pyrites or as marcasites, both of which have the same chemical formula. The former usually predominates. Free sulfur is occasionally found and also small quantities of sulfates. The organic sulfur had its origin in the protein of the plant and animal life of which coal is the degradation product, according to the theory discussed in Chapter I under the subject of the origin of coal. Powell and Parr (223) examined a number of different coals to ascertain the distribution of sulfur between inorganic and organic compounds, They found sulfur in both the resinic and organic bodies, whose proximate analyses were given in Table 4, and whose extraction has been discussed in Chapter I under the subject of destructive distillation. On the average, the sulfur of coal is about equally distributed between organic and inorganic compounds. Table 99 gives the distribution of sulfur among different classes of compounds as determined by Powell and Parr (223). All the specimens were of Illinois coal except Sample #5, which was form Tennessee.
The variation of the amount of sulfate which was present in the various samples is interesting, in view of the different ages of the coals after mining. During storage, oxidation of the coal, as a whole, includes oxidation of the sulfur compounds, and the iron pyrites may oxidize to such an extent that, after 2 or 3 years standing, sulfate may become the major sulfur compound. The sulfur content of Sample #3 in Table 99 contained less than 0.01% sulfate, when freshly mines, as compared with 1.31% after standing 2 years in a flask.

When coal is carbonized, the sulfur divides between the volatile products and the solid residuum. The ratio of volatile to residual sulfur varies between wide limits, but remains fairly constant for a given coal. The factors underlying the volatility of the sulfur are not well known. Undoubtedly, certain constituents of the coal, other than the sulfur, have a marked influence in this respect, but probably the most decisive factor is the relative amounts of the different sulfur compounds that are present in the coal. The sulfur compounds in the volatile matter from distillation are largely in the form of hydrogen sulfide, or thiophene derivates, while the residual sulfur in the coke is principally of an unknown organic nature with traces of sulfides. When the coke from coal Sample #4 of Table 99 was examined by Powell and parr (223) they found that 1.37% sulfur remained, of which 0.3% was sulfide and the rest organic. They found that the sulfate was entirely reduced, not to sulfide, but probably to form unknown organic sulfur compounds. The pyretic sulfur was totally decomposed, part being volatilized and part remaining as sulfide. Probably the resinic sulfur of the coal was left in the coke, but in a different form, while the humous sulfur was partly volatilized and partly left as residuum of changed form.
Powell (224) extensively investigated the reaction of coal sulfur in the coking process. When mineralogical pyrite is heated, very little decomposition is observed at 500° C, but at 1000° C it is entirely decomposed into equal proportions of free sulfur and sulfide sulfur. If moisture or other hydrogen-yielding compounds are present, the free sulfur will be partially converted to hydrogen sulfide. When mineralogical pyrite is mixed with an equal amount of coal and heated to the same temperature, it is found that half of the total sulfur appears as ferrous sulfide, about half as much again is evolved as hydrogen sulfide, while the remainder is divided in the ratio of about two to one as free and organic sulfur, respectively. It has been demonstrated by Campbell (225) and verified by Powell (224) that the ferrous sulfide residuum is pyrrhotite, or magnetic sulfide of iron, which is not a definite chemical compound, but a solid solution of sulfur in ferrous sulfide.
According to Powell (224), the total sulfur in the coal is the most important factor bearing on the sulfur content of the coke and a careful scrutiny of the percentage of organic and inorganic sulfur contents of the raw coal did not reveal any constant relation to the sulfur content of the coke produced therefrom. This practically nullifies any statement as to the relative importance of removing organic and pyretic sulfur before carbonization.
The transformation of sulfur compounds at various coking temperatures was studied by Powell (224), who used a number of different coals. The results of his heating a Tennessee coal containing 4.25% sulfur are given in Table 100, from a critical study of which it is possible to draw a number of conclusions regarding the reactions undergone by the sulfur compounds during coking. From Table 100 it is seen that the pyretic sulfur is rapidly decomposed between 400° C and 500° C and that the sulfates, which have gradually disappeared as the temperature increases, are almost entirely decomposed at 500° C. As the temperature advances, there is a gradual increase of sulfide sulfur in the coke and there is a gradually increasing proportion of volatile sulfur compounds found in the gas and tar. At first, the organic sulfur present in the coal varies inversely as the temperature, but between 400° C and 500° C a radical change takes place with the formation of increasing amounts of organic sulfur as the temperature of carbonization is further advanced. It is interesting to note from Table 100 that from the region of low temperature distillation to the region of high temperature carbonization, the only sulfur remaining in the coke is in the form of sulfides or of organic compounds.


It should be noted that, starting with an initial sulfur content of 4.25% at 500° C, 2.95% sulfur remained in the coke and this was further reduced to 2.65% at 1000° C. Gentry (226) has pointed out that the high temperature zone in processes of low temperature carbonization by partial gasification is largely responsible for removal of sulfur in that type of retort, as compared with the ordinary externally heated designs. At the same time, there is an increase in sulfur present in the tar and gas to counterbalance the decrease in this constituent of the coke.
When Powell (224) heated Pocahontas, a West Virginia low sulfur bituminous coal, he found the initial sulfur, consisting of 0.08% pyritic, 0.01% sulfate, and 0.47% organic sulfur, was transformed to a residuum with 0.01% pyretic, 0.43% organic, and 0.04% sulfide sulfur, 0.06% of the sulfur volatilizing as hydrogen sulfide, and 0.02% distilling into the tar. At 1000° C, 0.27% organic and 0.09% sulfide sulfur was found in the solid residuum, while 0.17% appeared as hydrogen sulfide and 0.03% was found in the tar. The difference in the transformations taking place among the sulfur compounds of Tennessee coal and of Pocahontas coal may be explained by the fact that the latter contained very little pyretic sulfur and practically no sulfate. Further results from the treatment of Pennsylvania coals from the Pittsburgh and Upper Freeport beds are reproduced in Table 101 principally for reference, as it will be observed that the transformations do not differ greatly from those taking place during the carbonization of Tennessee bituminous coal.
Powell (224) next submitted a mixture of Joliet coking coal to study. This consisted of a mixture of 65% Pocahontas, 20% Kentucky, and 15% washed Illinois coals. The sulfur, which was present in the raw mixture, amounted to 0.82%, distributed as 0.26% pyretic and 0.56% organic sulfur. After carbonization at 500° C, the sulfur in the coke consisted of 0.12% pyretic, 0.44% organic, and 0.08% sulfide, while the volatile products contained sulfur to the extent of 0.02% in the tar and 0.16% as hydrogen sulfide in the gas. At 1000° C, the residual sulfur was distributed as 0.49% organic and 0.06% sulfide, while the volatile sulfur consisted of 0.02% in the tar and 0.25% as hydrogen sulfide. The general transformations in this mixed coking coal are the same as those already discussed.
When an Indiana coal containing 1.38% sulfur, distributed as 0.7% pyretic, 0.03% sulfate, and 0.65% organic, was washed, the total sulfur then amounted to 1.18%, allocate as 0.25% pyretic, 0.03% sulfate, and 0.9% organic. Powell (224), in comparing the results with washed and unwashed Indiana coal, concluded first that in the raw coal, where the inorganic sulfur predominated, a larger quantity of metallic sulfides are converted into the organic form than in the washed coal, and second, that in the washed coal, where the organic sulfur predominated, more of the organic sulfur is decomposed into hydrogen sulfide than in the raw coal.
These extensive investigations on the transformation of sulfur constituents of coal during carbonization establish 5 classes of primary reactions. First, there is the complete decomposition of the pyrite and marcasite to ferrous sulfide, pyrrhotite, and hydrogen sulfide. This reaction reaches its maximum between 400° C and 500° C, but begins at about 300° C and is complete at about 600° C. Second, there is the reduction of sulfates to sulfides, which is complete at 500° C. Third, the organic sulfur decomposes to form hydrogen sulfide. Fourth, a small part of the organic sulfur decomposes to form volatile sulfur compounds, which are collected in the tar. Fifth, a portion of the ferrous sulfide and pyrrhotite that is formed apparently enters into combination with the carbon to form organic sulfur compounds in the neighborhood of 500° C. his last reaction has also been noted by Parr (227). In addition to these 5 primary reactions, there are a few additional secondary reactions, such as the formation of hydrogen sulfide by the attack of hydrogen in the gas on the organic sulfur compounds and the reduction of hydrogen sulfide by red-hot coke to form carbon disulfide. Lewes (17) has already shown that carbon disulfide is never a primary decomposition product of the distillation of coal and Powell (224) concurs in this conclusion.
Powell (224) has shown that from one-fourth to one-half of the coal sulfur can be removed by washing and that there is a corresponding reduction of sulfur in the coke. However efficient this method of processing, Fraser and Yancey (228) pointed out that this washing removes only a part of the sulfur that is present as pyrites, but does not eliminate the finely disseminated pyrite nor the organic sulfur. Besides washing, a number of methods have been proposed to desulfurize coke, all of which involve the elimination of sulfur compounds as volatile components or conversion of the sulfur to soluble forms which may be leached. To the first class belong such proposals as the introduction of steam, air, chlorine, carbon monoxide, and hydrogen during coking, while the second class involves the use of such addition agents as sodium chloride, sodium carbonate, and manganese dioxide. Most of these schemes, however, have never become commercial because of the cost or of inefficiency in sulfur reduction. It has been pretty well shown, in the foregoing tables, that most of the sulfur remaining in the coke is inorganic in nature, so that any method of desulfurization must attack the sulfur-carbon compounds.
Powell (229), noting the quantitative method for sulfur determination of Oteha (230), in which nascent hydrogen was used to attack the sulfur, studied the elimination of sulfur from coke by passing hydrogen through the coking mass. His first experiments were made on Pittsburgh coal containing 1.72% sulfur, distributed as shown in Figure 101. This coal was first coked at 1000° C for 2 hours until no more sulfur was removed by carbonization, at which time the residuum contained 1.9% sulfur, or 1.14% referred to the raw coal. Pure hydrogen was then passed through the furnace for one hour, after which the coke sulfur was reduced to 1.55%, or 0.93% referred to the raw coal. A second experiment was made with the same coal by introducing the hydrogen from the beginning of carbonization. In this test the coal was heated at 500° C for 2 hours and then at 1000° C for 2 hours with the result that the sulfur of the coke was reduced to 0.86%, or 0.52% referred to the coal.
The Upper Freeport coal of Table 101, containing 1.21 sulfur, was also treated. When this coal was treated with hydrogen for 2 hours at 500° C, it was found that the coal pyrite was completely decomposed, whereas only about half had been destroyed without hydrogen, according to Table 101. The speed of pyrite decomposition depends not only upon the temperature but upon the partial pressure of sulfur over it, so that the presence of hydrogen reduces this partial pressure, the decomposition becomes more rapid. As Table 101 shows that the pyretic sulfur would be destroyed in any event by thermal decomposition, the presence of hydrogen does not effect removal of this constituent from the coke, but merely permits its transformation to become complete at a lower temperature. When the Upper Freeport coal was treated at 500° C for one hour and then at 1000° C for one hour with slightly moist hydrogen, it was found that the coke contained 0.82% of the original sulfur after the 500° C stage and only 0.07% after the 1000° C stage. The distribution of sulfur after the 500° C stage was 0.01% pyretic, 0.01% sulfate, 0.63% organic, 0.17% sulfide, 0.36% evolved as hydrogen sulfide, and 0.03% in the tar. These data should be compared with those in Table 101, which gives the sulfur distribution at the same temperature for the same coal when hydrogen was not used. Although the sulfur reduction at 1000° C amounted to over 90%, the passage of hydrogen produced no effect on the character of the coke, which was of a rather fragile nature to begin with. The dsulfurizing action of hydrogen is due to its conversion of the organic sulfur compounds to sulfuretted hydrogen, which action is more vigorous at the higher temperatures. Monkhouse and Cobb (221), however, claim that when the pyretic sulfur is reduced by heat to ferrous sulfide, it is not further necessary for the sulfur to form a carbon compound, but that it can be decomposed freely at 1000° C by hydrogen, the statement of Roscoe and Schorlemmer (231), as quoted by Powell (229), to the contrary nothwithstanding.

Figure 52 shows the amount of sulfur removed from Upper Freeport coal as a function of time, as determined by Powell (229). The illustration shows the results of purging with pure hydrogen gas, as well as with a by-produce coal gas which was rich in hydrogen. For comparison, curves showing the elimination of sulfur as it ordinarily occurs during coking in by-product ovens and in primary distillation are given. Three hours of purging with by-product gas reduced the coke sulfur from 1.21% to 0.34%. When the Joliet coking coal mixture, already discussed in this chapter under the subject of temperature effect, was simultaneously coked and purged with hydrogen for 3 hours, the coke residuum contained but 0.29% sulfur, as compared with 0.75% sulfur when hydrogen was not used.
Monkhouse and Cobb (221), while studying the liberation of nitrogen as ammonia from coal and coke, also determined the liberation of sulfur as hydrogen sulfide. Analyses of the coke ash has already been given under the discussion of the effect of nitrogenous and hydrogenous atmospheres on the oxidation and dissociation of ammonia. Analyses of raw coal and the 500° C, 800° C and 1100° C cokes which were tested were also given in Table 93, from which it is seen that the higher the temperature of carbonization, the less the percentage of initial sulfur that remains in the coke. Taking the sulfur in the coal as 100%, that in the 500° C coke and in the 800° C coke was 56.4% and 50.1% respectively.
Figure 53 shows the result of treating 500° C low temperature coke successively with nitrogen, hydrogen, and steam at 800° C. Comparison of this illustration with that of Figure 49, for the same set of experiments in the removal of hydrogen, sows a great similarity. In an atmosphere of nitrogen, the removal of sulfur is practically negligible, but in atmospheres of hydrogen and of steam the reaction of the sulfur to form hydrogen sulfide is far more sensitive than that of nitrogen to form ammonia. The use of nitrogen removed 1% of the coke sulfur and the hydrogen removed about 51.8%of the coke sulfur, whereas treatment with steam to complete gasification removed an additional 40.2% of the total sulfur in the coke, amounting in all to 93%, with the rest remaining in the ash and unaccounted for. When the experiment was repeated with omission of the hydrogen stage, only 53.7% of the coke sulfur was removed as hydrogen sulfide. When the hydrogen stage was omitted, apparently quite apportion of the coke sulfur was oxidized, the hydrogen sulfide and sulfur dioxide later reacting to precipitate sulfur as a white milk in the gas washers.



Figure 54 shows the result of heating the 500° C low temperature coke to various temperatures in an atmosphere of hydrogen. A similar experiment up to 800° C in an atmosphere of nitrogen resulted in only 0.8% of the sulfur of the coke being liberated as hydrogen sulfide. Up to 800° C, the evolution of hydrogen sulfide from the low temperature coke in a hydrogenous atmosphere was fairly steady, but at 800° C it fell off rapidly. At 900° C, however, there was a rapid hydrogenation of the sulfur which fell off after about 20 hours treatment and could not be materially increased by raising the temperature to even 1000° C, Treatment in stages of 100° C, up to 800° C, removed 63.4% of the coke sulfur as hydrogen sulfide in an atmosphere of hydrogen, as compared with only 52.8% when it was heated to 800° C final temperature in a single heating stage, as seen form Figure 53. After 200 hours of treatment and 1000° C final temperature, 93.8% of the sulfur was removed from the coke as hydrogen sulfide. When next an atmosphere of steam was used in multi-stage heating of the 500° C coke, only 66.4% of the sulfur was finally obtained as hydrogen sulfide and only traces of sulfur were left in the ash, the remaining sulfur being unaccounted for. When the 1100° C coke was heated up to 1000° C in nitrogen and steam, only 60.6% of the sulfur was evolved as hydrogen sulfide, although all was driven from the coke, two-fifths of that evolved appearing up to 800° C, three-fifths at 900° C, and only a fraction of 1% at 1000° C.
The result of heating the 800° C coke up to 1000° C in stages with an atmosphere of hydrogen is shown in Figure 55 and Table 102 gives a sulfur balance for these conditions, as well as for an atmosphere of nitrogen. From the sulfur balance and the illustration, it is patent that in a nitrogenous atmosphere very little sulfur in any form was eliminated from the coke. At 600° C in an atmosphere of hydrogen very little hydrogen sulfide was removed from the 800° C coke, but at reheat temperatures of 800° C and 1000° C, hydrogen sulfide was evolved at a fairly uniform rate for 8-hour periods of heating.


When coke, which was produced at 1000° C, was heated through these temperature stages in a hydrogenous atmosphere, the results noted in Figure 56 were obtained. Only 25.3% of the sulfur in the coke appeared as hydrogen sulfide when heated for over 16 hours to a final temperature of 1000° C, about 76% remaining in the coke. However, when steam was mixed with the hydrogen and passed over the coke, decidedly different results were obtained, as shown in Figure 56. With the moist hydrogen atmosphere, very little hydrogen sulfide was produced at 600° C, as before, but when the temperature was raised to 800° C, the evolution became rapid, decreasing after about 5 hours, but hydrogen sulfide was evolved in quantities, even beyond 12 hours of heating. Finally, when the temperature was increased to 1000° C, the evolution of hydrogen sulfide became voluminous, the elimination of sulfur being at the rate of about 11% of that in the coke per hour of treatment.
Monkhouse and Cobb (220, 221) noted two important differences in the liberation of ammonia and of hydrogen sulfide from coke by hydrogen. In the first place, there was no loss of hydrogen sulfide by thermal dissociation, as in the case of ammonia, and in the second place, hydrogen at 800° C attacked the sulfur compounds in 1100° C hard coke, but not the nitrogen compounds. The first of these differences is explained by the fact that the dissociation equilibrium of hydrogen sulfide is only 5.5% dissociated at 750° C, 15.6% dissociated at 945° C, and 30.7% dissociated at 1132° C.
The Civilization Kit
... It's Your Best Bet & Investment in Sustainable Humanity on Earth ...
Everything @ rexresearch.com on a Data DVD
ORDER PAGE